
 French
French Deutsch
DeutschAcide désoxyribonucléique — Wikipédia
« ADN » redirige ici. Pour les autres significations, voir ADN (homonymie).
Pour les articles homonymes, voir DNA.




L'acide désoxyribonucléique, ou ADN, est une macromolécule biologique présente dans presque toutes[a] les cellules ainsi que chez de nombreux virus. L'ADN contient toute l'information génétique, appelée génome, permettant le développement, le fonctionnement et la reproduction des êtres vivants. C'est un acide nucléique, au même titre que l'acide ribonucléique (ARN). Les acides nucléiques sont, avec les peptides et les glucides, l'une des trois grandes familles de biopolymères essentiels à toutes les formes de vie connues.
Les molécules d'ADN des cellules vivantes sont formées de deux brins antiparallèles enroulés l'un autour de l'autre pour former une double hélice. On dit que l'ADN est bicaténaire, ou double brin. Chacun de ces brins est un polymère appelé polynucléotide. Chaque monomère qui le constitue est un nucléotide, lequel est formé d'une base nucléique, ou base azotée — adénine (A), cytosine (C), guanine (G) ou thymine (T) — liée à un ose — ici, le désoxyribose — lui-même lié à un groupe phosphate. Les nucléotides polymérisés sont unis les uns aux autres par des liaisons covalentes entre le désoxyribose d'un nucléotide et le groupe phosphate du nucléotide suivant, formant ainsi une chaîne où alternent oses et phosphates, avec des bases nucléiques liées chacune à un ose. L'ordre dans lequel se succèdent les nucléotides le long d'un brin d'ADN constitue la séquence de ce brin. C'est cette séquence qui porte l'information génétique. Celle-ci est structurée en gènes, qui sont exprimés à travers la transcription en ARN. Ces ARN peuvent être non codants — ARN de transfert et ARN ribosomique notamment — ou bien codants : il s'agit dans ce cas d'ARN messagers, qui sont traduits en protéines par des ribosomes. La succession des bases nucléiques sur l'ADN détermine la succession des acides aminés qui constituent les protéines issues de ces gènes. La correspondance entre bases nucléiques et acides aminés est le code génétique. L'ensemble des gènes d'un organisme constitue son génome.
Les bases nucléiques d'un brin d'ADN peuvent interagir avec les bases nucléiques d'un autre brin d'ADN à travers des liaisons hydrogène, qui déterminent des règles d'appariement entre paires de bases : l'adénine et la thymine s'apparient au moyen de deux liaisons hydrogène, tandis que la guanine et la cytosine s'apparient au moyen de trois liaisons hydrogène. Normalement, l'adénine et la cytosine ne s'apparient pas, tout comme la guanine et la thymine. Lorsque les séquences des deux brins sont complémentaires, ces brins peuvent s'apparier en formant une structure bicaténaire hélicoïdale caractéristique qu'on appelle double hélice d'ADN. Cette double hélice est bien adaptée au stockage de l'information génétique : la chaîne oses-phosphates est résistante aux réactions de clivage ; de plus, l'information est dupliquée sur les deux brins de la double hélice, ce qui permet de réparer un brin endommagé à partir de l'autre brin resté intact ; enfin, cette information peut être copiée à travers un mécanisme appelé réplication de l'ADN au cours duquel une double hélice d'ADN est recopiée fidèlement en une autre double hélice portant la même information. C'est en particulier ce qui se passe lors de la division cellulaire : chaque molécule d'ADN de la cellule mère est répliquée en deux molécules d'ADN, chacune des deux cellules filles recevant ainsi un jeu complet de molécules d'ADN, chaque jeu étant identique à l'autre.
Dans les cellules, l'ADN est organisé en structures appelées chromosomes. Ces chromosomes ont pour fonction de rendre l'ADN plus compact à l'aide de protéines, notamment d'histones, qui forment, avec les acides nucléiques, une substance appelée chromatine. Les chromosomes participent également à la régulation de l'expression génétique en déterminant quelles parties de l'ADN doivent être transcrites en ARN. Chez les eucaryotes (animaux, plantes, champignons et protistes), l'ADN est essentiellement contenu dans le noyau des cellules, avec une fraction d'ADN présent également dans les mitochondries ainsi que, chez les plantes, dans les chloroplastes. Chez les procaryotes (bactéries et archées), l'ADN est contenu dans le cytoplasme. Chez les virus qui contiennent de l'ADN, celui-ci est stocké dans la capside. Quel que soit l'organisme considéré, l'ADN est transmis au cours de la reproduction : il joue le rôle de support de l'hérédité. La modification de la séquence des bases d'un gène peut conduire à une mutation génétique, laquelle peut, selon les cas, être bénéfique, sans conséquence ou néfaste pour l'organisme, voire incompatible avec sa survie. À titre d'exemple, la modification d'une seule base d'un seul gène — celui de la β-globine, une sous-unité protéique de l'hémoglobine A — du génotype humain est responsable de la drépanocytose, une maladie génétique parmi les plus répandues dans le monde.
Définition
[modifier | modifier le code]L'ADN « permet de constituer, par un enroulement en double hélice, des molécules de grande taille (macromolécules) constituant ainsi les chromosomes qui codent l'information génétique des êtres vivants »[1].
Propriétés générales
[modifier | modifier le code]

L'ADN est un long polymère formé par la répétition de monomères appelés nucléotides. Le premier ADN a été identifié et isolé en 1869 à partir du noyau de globules blancs par le Suisse Friedrich Miescher. Sa structure en double hélice a été mise en évidence en 1953 par le Britannique Francis Crick et l'Américain James Watson à partir des données expérimentales obtenues par les Britanniques Rosalind Franklin (en fait volées à cette dernière) et Maurice Wilkins (qui les a communiquées). Cette structure, commune à toutes les espèces, est constituée de deux chaînes polynucléotidiques hélicoïdales enroulées l'une autour de l'autre autour d'un axe commun, avec un pas d'environ 3,4 nm pour un diamètre d'environ 2,0 nm[2]. Une autre étude mesurant les paramètres géométriques de l'ADN en solution donne un diamètre de 2,2 à 2,6 nm avec une longueur par nucléotide de 0,33 nm[3]. Bien que chaque nucléotide soit très petit, les molécules d'ADN peuvent en contenir des millions et atteindre des dimensions significatives. Par exemple, le chromosome 1 humain, qui est le plus grand des chromosomes humains, contient environ 220 millions de paires de bases[4] pour une longueur linéaire de plus de 7 cm.
Dans les cellules vivantes, l'ADN n'existe généralement pas sous forme monocaténaire (simple brin) mais plutôt sous forme bicaténaire (double brin) avec une configuration en double hélice[2]. Les monomères constituant chaque brin d'ADN comprennent un segment de la chaîne désoxyribose–phosphate et une base nucléique liée au désoxyribose. La molécule résultant de la liaison d'une base nucléique à un ose est appelée nucléoside ; l'adjonction d'un à trois groupes phosphate à l'ose d'un nucléoside forme un nucléotide. Un polymère résultant de la polymérisation de nucléotides est appelé polynucléotide. L'ADN et l'ARN sont des polynucléotides.
L'ose constituant le squelette de la molécule est le 2’-désoxyribose, dérivé du ribose. Ce pentose alterne avec des groupes phosphate en formant des liaisons phosphodiester entre les atomes no 3’ et no 5’ de résidus de désoxyribose adjacents[5]. En raison de cette liaison asymétrique, les brins d'ADN ont un sens. Dans une double hélice, les deux brins d'ADN sont de sens opposés : ils sont dits antiparallèles. Le sens 5’ vers 3’ d'un brin d'ADN désigne conventionnellement celui de l'extrémité portant un groupe phosphate –PO32− vers l'extrémité portant un groupe hydroxyle –OH ; c'est dans ce sens qu'est synthétisé l'ADN par les ADN polymérases. L'une des grandes différences entre l'ADN et l'ARN est le fait que l'ose du squelette de la molécule est le ribose dans le cas de l'ARN à la place du désoxyribose de l'ADN, ce qui joue sur la stabilité et la géométrie de cette macromolécule.
La double hélice d'ADN est stabilisée essentiellement par deux forces : les liaisons hydrogène entre nucléotides d'une part, et les interactions d'empilement des cycles aromatiques des bases nucléiques d'autre part[6]. Dans l'environnement aqueux de la cellule, les liaisons π conjuguées de ces bases s'alignent perpendiculairement à l'axe de la molécule d'ADN afin de minimiser leurs interactions avec la couche de solvatation et, par conséquent, leur enthalpie libre. Les quatre bases nucléiques constitutives de l'ADN sont l'adénine (A), la cytosine (C), la guanine (G) et la thymine (T), formant respectivement les quatre nucléotides suivants, composant l'ADN :




Classification et appariement des bases nucléiques
[modifier | modifier le code]Les quatre bases nucléiques de l'ADN sont de deux types : d'une part les purines — adénine et guanine — qui sont des composés bicycliques comprenant deux hétérocycles à cinq et six atomes respectivement, d'autre part les pyrimidines — cytosine et thymine — qui sont des composés monocycliques comprenant un hétérocycle à six atomes. Les paires de bases de la double hélice d'ADN sont constituées d'une purine interagissant avec une pyrimidine à travers deux ou trois liaisons hydrogène :
- une adénine interagit avec une thymine à travers deux liaisons hydrogène ;
- une guanine interagit avec une cytosine à travers trois liaisons hydrogène.
En raison de cette complémentarité, toute l'information génétique portée par l'un des brins de la double hélice d'ADN est également portée à l'identique sur l'autre brin. C'est sur ce principe que repose le mécanisme de la réplication de l'ADN, et c'est sur cette complémentarité entre bases nucléiques que reposent toutes les fonctions biologiques de l'ADN dans les cellules vivantes.






L'ADN de certains virus, tels que les bactériophages PBS1 et PBS2 de Bacillus subtilis, le bactériophage φR1-37 de Yersinia[7] et le phage S6 de Staphylococcus[8], peut remplacer la thymine par l'uracile, une pyrimidine habituellement caractéristique de l'ARN mais normalement absente de l'ADN, où on ne le trouve que comme produit de dégradation de la cytosine.
Appariements non canoniques entre bases nucléiques
[modifier | modifier le code]Les bases nucléiques s'apparient le plus souvent en formant les paires de bases dites « Watson-Crick » correspondant à deux ou trois liaisons hydrogène établies entre deux bases orientées anti sur les résidus de désoxyribose. Des liaisons hydrogène peuvent cependant également s'établir entre une purine orientée syn et une pyrimidine orientée anti : il s'agit dans ce cas d'un appariement Hoogsteen. Une paire de bases Watson-Crick est susceptible d'établir en plus des liaisons hydrogène de type Hoogsteen avec une troisième base, ce qui permet la formation de structures à trois brins d'ADN.
-
 Appariements AT et GC : Watson-Crick (à gauche) et Hoogsteen (à droite).
Appariements AT et GC : Watson-Crick (à gauche) et Hoogsteen (à droite). -
 Appariements Hoogsteen et Watson-Crick entre trois bases.
Appariements Hoogsteen et Watson-Crick entre trois bases.


Sens, antisens et ambisens
[modifier | modifier le code]Seul l'un des brins d'un segment d'ADN constituant un gène est transcrit en ARN fonctionnel, de sorte que les deux brins d'un gène ne sont pas équivalents : celui qui est transcrit en ARN fonctionnel est dit à polarité négative et porte une séquence antisens, tandis que le brin complémentaire — qui peut également être transcrit en ARN, mais non fonctionnel — est dit à polarité positive et porte une séquence d'ADN sens. Le brin transcrit en ARN fonctionnel est parfois appelé brin codant, mais cette désignation n'est valable qu'au sein d'un gène donné car les deux brins d'une même double hélice d'ADN peuvent coder différentes protéines ; on parle alors de brins ambisens[9],[10],[11]. Des ARN sont également transcrits à partir des séquences d'ADN sens — avec par conséquent des séquences d'ARN antisens — aussi bien chez les procaryotes que chez les eucaryotes, mais leur rôle biologique n'est pas entièrement élucidé[12] ; l'une des hypothèses est que ces ARN antisens pourraient intervenir dans la régulation de l'expression génétique à travers l'appariement entre séquences d'ARN sens et antisens, qui sont, par définition, complémentaires[13].
La distinction entre brins d'ADN sens et antisens est brouillée dans certains types de gènes chevauchants, assez rares chez les procaryotes et les eucaryotes mais plus fréquents sur les plasmides et chez les virus, dans lesquels les deux brins d'un même segment d'ADN encodent chacun un ARN fonctionnel différent[14]. Chez les bactéries, ce chevauchement peut jouer un rôle dans la régulation de la transcription des gènes[15] tandis que, chez les virus, les gènes chevauchants accroissent la quantité d'information génétique susceptible d'être encodée dans la petite taille du génome viral[16].
Supertours et surenroulement
[modifier | modifier le code]L'ADN relâché peut être linéaire, comme c'est typiquement le cas chez les eucaryotes, ou circulaire, comme chez les procaryotes. Il peut cependant être entortillé de façon parfois complexe sous l'effet de l'introduction de tours d'hélice supplémentaires ou de la suppression de tours dans la double hélice. La double hélice d'ADN ainsi surenroulée sous l'effet de supertours positifs ou négatifs présente un pas respectivement raccourci ou allongé par rapport à son état relâché : dans le premier cas, les bases nucléiques sont arrangées de façon plus compacte ; dans le second cas, elles interagissent au contraire de façon moins étroite[17]. In vivo, l'ADN présente généralement un surenroulement légèrement négatif sous l'effet d'enzymes appelées ADN topoisomérases[18], qui sont également indispensables pour relâcher les contraintes introduites dans l'ADN lors des processus qui impliquent que la double hélice soit déroulée pour en séparer les deux brins, comme c'est notamment le cas lors de la réplication de l'ADN et lors de sa transcription en ARN[19].
Propriétés physicochimiques de la double hélice
[modifier | modifier le code]Les liaisons hydrogène n'étant pas des liaisons covalentes, elles peuvent être rompues assez facilement. Il est ainsi possible de séparer les deux brins de la double hélice d'ADN à la façon d'une fermeture à glissière aussi bien mécaniquement que sous l'effet d'une température élevée[20], ainsi qu'à faible salinité, à pH élevé — solution basique — et à pH faible — solution acide, qui altère cependant l'ADN notamment par dépurination. Cette séparation des brins d'un ADN bicaténaire pour former deux molécules d'ADN monocaténaires est appelé fusion ou dénaturation de l'ADN. La température à laquelle 50 % de l'ADN bicaténaire est dissocié en deux molécules d'ADN monocaténaire est dite température de fusion ou température de semi-dénaturation de l'ADN, notée Tm. On peut la mesurer en suivant l'absorption optique à 260 nm de la solution contenant l'ADN : cette absorption augmente au cours du désappariement, ce qu'on appelle hyperchromicité. Les molécules d'ADN monocaténaire libérées n'ont pas de configuration particulière, mais certaines structures tridimensionnelles sont plus stables que d'autres[21].
La stabilité d'une double hélice d'ADN dépend essentiellement du nombre de liaisons hydrogène à briser pour en séparer les deux brins. Par conséquent, plus la double hélice est longue, plus elle est stable. Cependant, les paires GC étant unies par trois liaisons hydrogène au lieu de deux pour les paires AT, la stabilité de molécules d'ADN bicaténaires de même longueur croît avec le nombre de paires GC qu'elles contiennent[22], mesuré par leur taux de GC. Cet effet est renforcé par le fait que les interactions d'empilement entre bases nucléiques d'un même brin d'ADN sont plus fortes entre résidus de guanine et de cytosine, de sorte que la séquence de l'ADN influence également sur sa stabilité. La température de fusion de l'ADN dépend par conséquent de la longueur des molécules, de leur taux de GC, de leur séquence, de leur concentration dans le solvant et de la force ionique dans celui-ci. En biologie moléculaire, on observe que les segments d'ADN bicaténaire dont la fonction implique que les deux brins de la double hélice puissent s'écarter facilement possèdent un taux élevé de paires AT[23] : c'est le cas de la séquence TATAAT typique de la boîte de Pribnow de certains promoteurs.
Géométrie de la double hélice
[modifier | modifier le code]


Les deux brins de l'ADN forment une double hélice dont le squelette détermine deux sillons. Ces sillons sont adjacents aux paires de bases et sont susceptibles de fournir un site de liaison pour diverses molécules. Les brins d'ADN n'étant pas positionnés de façon symétrique par rapport à l'axe de la double hélice, ils définissent deux sillons de taille inégale : le grand sillon est large de 1,2 nm tandis que le petit sillon est large de 0,6 nm[24]. Les bords des bases nucléiques sont plus accessibles dans le grand sillon que dans le petit sillon. Ainsi, les protéines, telles que les facteurs de transcription, qui se lient à des séquences spécifiques dans l'ADN bicaténaire le font généralement au niveau du grand sillon[25].
Il existe de nombreux conformères possibles de la double hélice d'ADN. Les formes classiques sont appelées ADN A, ADN B et ADN Z, dont seules les deux dernières ont été observées directement in vivo[5]. La conformation adoptée par l'ADN bicaténaire dépend de son degré d'hydratation, de sa séquence, de son taux de surenroulement, des modifications chimiques des bases qui le composent, de la nature et de la concentration des ions métalliques en solution, voire de la présence de polyamines[26].
- L'ADN B est la forme la plus courante de la double hélice dans les conditions physiologiques des cellules vivantes[27]. Il ne s'agit cependant pas d'une conformation définie par des paramètres géométriques stricts mais plutôt d'un ensemble de conformations apparentées survenant aux niveaux d'hydratation élevés observés dans les cellules vivantes. Leur étude par cristallographie aux rayons X révèle des diagrammes de diffraction et de diffusion caractéristiques de paracristaux moléculaires très désordonnés[28]. La forme B est une hélice droite avec des paires de bases perpendiculaires à l'axe de l'hélice passant au centre de l'appariement de ces dernières. Un tour d'hélice a une longueur d'environ 3,4 nm et contient en moyenne 10,4 à 10,5 paires de bases, soit environ 21 nucléotides, pour un diamètre de l'ordre de 2,0 nm. Les bases sont orientées en position anti sur les résidus de désoxyribose, lesquels présentent un plissement endocyclique C2’-endo du cycle furanose. Les deux sillons de cette configuration ont une largeur typique de 2,2 nm pour le grand et de 1,2 nm pour le petit[24].
- L'ADN A s'observe dans les échantillons d'ADN plus faiblement hydraté, à force ionique plus élevée, en présence d'éthanol ainsi qu'avec les hybrides bicaténaires d'ADN et d'ARN. Il s'agit d'une double hélice droite dont l'axe ne passe plus par les paires de bases. Cette double hélice est plus large, avec un diamètre de l'ordre de 2,3 nm mais un pas de seulement 2,8 nm pour 11 paires de bases par tour d'hélice. Les bases elles-mêmes demeurent orientées en position anti sur les résidus de désoxyribose, mais ces derniers présentent un plissement endocyclique C3’-endo.
- L'ADN Z est plus contraint que les formes A et B de l'ADN et s'observe préférentiellement dans les régions riches en paires guanine–cytosine lors de la transcription de l'ADN en ARN. Il s'agit d'une double hélice gauche, dont l'axe s'écarte significativement des paires de bases. Cette double hélice est plus étroite, avec un diamètre d'environ 1,8 nm et un pas d'environ 4,5 nm pour 12 paires de bases par tour d'hélice. Les pyrimidines sont orientées en position anti sur les résidus de désoxyribose, dont le cycle furanose possède en leur présence un plissement C2’-endo, tandis que les purines sont orientées en position syn sur des résidus de désoxyribose qui possède en leur présence un plissement endocyclique C2’-exo. La forme Z de l'ADN serait notamment provoquée in vivo par une enzyme appelée ADAR1[29],[30],[31].

| Paramètre | ADN A | ADN B | ADN Z |
|---|---|---|---|
| Sens de l'hélice | droite | droite | gauche |
| Motif répété | 1 bp | 1 bp | 2 bp |
| Angle de rotation par paire de bases | 32,7° | 34,3° | 60°/2 |
| Nombre de paires de bases par tour d'hélice | 11 | 10,5 | 12 |
| Pas de l'hélice par tour | 2,82 nm | 3,32 nm | 4,56 nm |
| Allongement de l'axe par paire de bases | 0,24 nm | 0,32 nm | 0,38 nm |
| Diamètre | 2,3 nm | 2,0 nm | 1,8 nm |
| Inclinaison des paires de bases sur l'axe de l'hélice | +19° | −1,2° | −9° |
| Torsion moyenne (propeller twist) | +18° | +16° | 0° |
| Orientation des substituants des bases sur les résidus osidiques | anti | anti | Pyrimidine : anti, Purine : syn |
| Plissement / torsion endocyclique du furanose (Sugar pucker) | C3’-endo | C2’-endo | Cytosine : C2’-endo, Guanine : C3’-endo |
Géométries particulières et configurations remarquables
[modifier | modifier le code]- Jonction de Holliday — Une jonction de Holliday est formée lors de la recombinaison homologue entre deux molécules d'ADN portant la même information génétique (chromosomes homologues, chromatides sœurs, etc.). Cette jonction présente une configuration cruciforme avec souvent des séquences symétriques qui lui permettent de se déplacer dans un sens ou dans l'autre. Sa résolution est effectuée par un complexe enzymatique appelé résolvase (EC ) et peut conduire à un enjambement entre les deux molécules, aboutissant à un échange de matériel génétique[35].
- ADN en épingle à cheveux — Les séquences d'ADN palindromiques peuvent se replier en formant des structures dites tige-boucle ou en épingle à cheveux[36]. Certaines répétitions, particulièrement les répétitions de trinucléotides (CAG)n ou (CTG)n peuvent former des épingles à cheveux imparfaites dans lesquelles les résidus de cytosine et de guanine sont appariés tandis que les résidus d'adénine ou de thymine ne le sont pas[37].
- ADN H ou ADN triplex — Cette forme d'ADN tricaténaire (triple brin) pourrait jouer un rôle dans la régulation fonctionnelle de l'expression des gènes en modulant leur transcription en ARN.
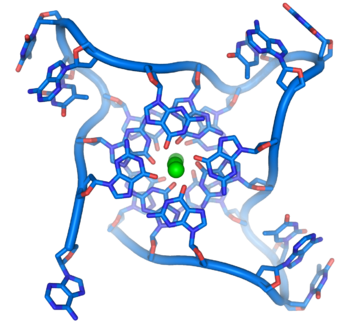
- ADN G ou G-quadruplex — L'extrémité des chromosomes linéaires est constitué d'une région spécialisée appelée télomère dont la fonction principale est de permettre la réplication de l'extrémité des chromosomes à l'aide d'une enzyme spécifique, la télomérase, dans la mesure où les enzymes qui répliquent l'ADN ne peuvent recopier l'extrémité 3' des chromosomes[38]. Ces structures protègent les extrémités des chromosomes et empêchent les systèmes de réparation de l'ADN de les considérer comme endommagées[39]. Dans les cellules humaines, les télomères sont généralement constitués d'ADN monocaténaire répétant des milliers de fois la simple séquence TTAGGG[40]. Ces séquences riches en résidus de guanine forment des « G-quartets » résultant de l'appariement Hoogsteen entre quatre résidus de guanine ; l'empilement de ces structures à quatre résidus de guanine forme un G-quadruplex à la structure particulièrement stable[41]. Ces structures sont également stabilisées par la chélation d'ions métalliques au centre de chaque G-quartet[42].
- ADN ramifié — L'ADN s'effile à une extrémité de la double hélice lorsque les séquences de ses deux brins cessent d'y être complémentaires : les deux brins s'écartent l'un de l'autre et la molécule adopte une configuration en Y. L'ADN peut alors se ramifier si un troisième brin possède une séquence susceptible de s'apparier avec les deux extrémités libres des deux brins effilés. Il se forme alors une structure en Y intégralement bicaténaire. Il est possible d'envisager des ramifications plus complexes avec de multiples branches[43].
-
![Jonction de Holliday (PDB 3CRX[44]).](//upload.wikimedia.org/wikipedia/commons/thumb/9/92/Holliday_junction_coloured.png/330px-Holliday_junction_coloured.png)
-
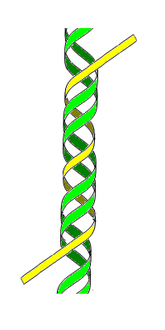 ADN H ou triplex
ADN H ou triplex
(3e brin en jaune) -
![Jonction de Holliday (PDB 3CRX[44]).](/fr/Fichier:Holliday_junction_coloured.png)


-
 ADN effilé
ADN effilé -
 ADN ramifié
ADN ramifié -
 Ramifications mutliples
Ramifications mutliples



Altérations chimiques
[modifier | modifier le code]Modifications des bases nucléiques
[modifier | modifier le code]
L'expression génétique de l'ADN dépend de la façon dont l'ADN est conditionné dans les chromosomes en une structure appelée chromatine. Certaines bases peuvent être modifiées lors de la formation de la chromatine, les résidus de cytosine des régions peu ou pas exprimées génétiquement étant généralement fortement méthylées, et ce majoritairement aux sites CpG. Les histones autour desquelles l'ADN est enroulé dans les chromatines peuvent également être modifiées de façon covalente. La chromatine elle-même peut être modifiée par des complexes de remodelage de la chromatine. De plus, la méthylation de l'ADN et la modification covalente des histones sont coordonnées pour affecter la chromatine et l'expression des gènes[45].
Ainsi, la méthylation des résidus de cytosine produit de la 5-méthylcytosine, qui joue un rôle important dans l'inactivation du chromosome X[46]. Le taux de méthylation varie entre organismes, le nématode Caenorhabditis elegans en étant totalement dépourvu tandis que les vertébrés ont environ 1 % de leur ADN contenant de la 5-méthylcytosine[47].
Les pyrimidines ont une structure moléculaire très similaire. Ainsi, la cytosine et la 5-méthylcytosine peuvent être désaminées pour produire l'uracile (qui n'est pas une base faisant partie du code de l'ADN) et la thymine, respectivement. La réaction de désamination pourrait par conséquent favoriser les mutations génétiques[48],[49].




Il existe également d'autres bases modifiées dans l'ADN, résultant par exemple de la méthylation de résidus d'adénine chez les bactéries[50] mais également chez des nématodes (Caenorhabditis elegans[51]), des algues vertes (Chlamydomonas[52]) et des drosophiles[53]. La 5-hydroxyméthylcytosine est un dérivé de la cytosine particulièrement abondant dans le cerveau des mammifères[54]. Des organismes tels que les flagellés Diplonema et Euglena et le genre Kinetoplastida, contiennent par ailleurs une pyrimidine glycosylée issue de l'uracile et appelée base J[55],[56] ; cette base modifiée agit comme signal de terminaison de transcription pour l'ARN polymérase II[57],[58]. Un certain nombre de protéines qui se lient spécifiquement à la base J ont été identifiées[59],[60],[61].
Lésions de la double hélice
[modifier | modifier le code]
L'ADN peut être endommagé par un grand nombre de mutagènes qui modifient sa séquence. Ces mutagènes comprennent les oxydants, les alkylants, les rayonnements électromagnétiques énergétiques tels que les ultraviolets et les rayons X et gamma, ainsi que les particules subatomiques des rayonnements ionisants tels que ceux résultant de la radioactivité voire des rayons cosmiques. Le type de dommages produits dépend du type de mutagène. Ainsi, les rayons ultraviolets sont susceptibles d'endommager l'ADN en produisant des dimères de pyrimidine en établissant des liaisons entre bases adjacentes d'un même brin d'ADN[63]. Les oxydants tels que les radicaux libres ou le peroxyde d'hydrogène produisent plusieurs types de dommages, comme des modifications de bases, notamment de la guanosine, et des cassures de la structure bicaténaire[64]. Une cellule humaine typique contient environ 150 000 bases endommagées par un oxydant[65]. Parmi ces lésions dues à des oxydants, les plus dangereuses sont les ruptures bicaténaires parce que ce sont les plus difficiles à réparer et qu'elles sont susceptibles de produire des mutations ponctuelles, des insertions et des délétions au sein de la séquence d'ADN, ainsi que des translocations chromosomiques[66]. Ces mutations sont susceptibles de provoquer des cancers. Les altérations naturelles de l'ADN, qui résultent par exemple de processus cellulaires produisant des dérivés réactifs de l'oxygène, sont assez fréquentes. Bien que les mécanismes de réparation de l'ADN résorbent l'essentiel de ces lésions, certaines d'entre elles ne sont pas réparées et s'accumulent au fil du temps dans les tissus postmitotiques des mammifères. L'accumulation de telles lésions non réparées semble être une importante cause sous-jacente du vieillissement[67],[68].
De nombreux mutagènes s'insèrent dans l'espace entre deux paires de bases adjacentes selon un mode qu'on appelle intercalation. La plupart des intercalations sont le fait de composés aromatiques et de molécules planes, telles que le bromure d'éthidium, les acridines, la daunorubicine ou la doxorubicine. Les bases doivent s'écarter afin de permettre l'insertion du composé d'intercalation, ce qui provoque une distorsion de la double hélice par désenroulement partiel. Ceci bloque à la fois la transcription et la réplication de l'ADN, entraînant cytotoxicité et mutations[69]. En conséquence, les composés d'intercalation peuvent être cancérogènes et, dans le cas du thalidomide, tératogènes[70]. D'autres composés tels que le benzo[a]pyrène diol époxyde et l'aflatoxine forment avec l'ADN des adduits qui provoquent des erreurs lors de la réplication[71]. Cependant, en raison de leur aptitude à bloquer la transcription et la réplication de l'ADN, d'autres toxines semblables sont également utilisées en chimiothérapie contre les cellules à prolifération rapide[72].
Fonctions biologiques
[modifier | modifier le code]

L'ADN se trouve essentiellement au sein de chromosomes, généralement linéaires chez les eucaryotes et circulaires chez les procaryotes. Chez ces derniers, il peut également se trouver en dehors des chromosomes, au sein de plasmides. L'ensemble de l'ADN d'une cellule constitue son génome. Le génome humain représente environ trois milliards de paires de bases distribués dans 46 chromosomes[73]. L'information contenue dans le génome est portée par des segments d'ADN formant les gènes. L'information génétique est transmise grâce aux règles spécifiques d'appariement des bases dites appariement Watson-Crick : les deux seules paires de bases normalement permises sont l'adénine avec la thymine et la guanine avec la cytosine. Ces règles d'appariement sont sous-jacentes aux différents processus à l'œuvre dans les fonctions biologiques de l'ADN :
- une double hélice d'ADN peut être répliquée en une autre double hélice en associant, à chaque base de chacun des brins, le nucléotide portant la base complémentaire selon les règles d'appariement de Watson-Crick : il se forme ainsi deux molécules d'ADN bicaténaire identiques là où il n'y en avait au départ qu'une seule, assurant la conservation de l'information au cours des cycles de vie successifs des cellules, ce qui fait de l'ADN le vecteur de l'hérédité ;
- l'information génétique d'une cellule est normalement conservée au cours du temps, mais peut être altérée par recombinaison ou par mutation, c'est-à-dire à la suite d'erreurs de réplication de l'ADN par addition, suppression ou substitution de nucléotides. Sous l'effet de la sélection naturelle, ces mutations constituent le moteur de l'évolution biologique des espèces ;
- enfin, l'information portée par l'ADN peut également être transcrite en ARN à travers la règle d'appariement de Watson-Crick qui, à chaque base de l'ADN, fait correspondre une et une seule base de l'ARN ; cet ARN est lui-même susceptible d'être à son tour traduit en protéines à travers le code génétique, selon un mécanisme de décodage, réalisé par des ARN de transfert, qui repose sur la même règle d'appariement entre bases nucléiques.
Réplication
[modifier | modifier le code]Lorsqu'une cellule se divise, elle doit répliquer l'ADN portant son génome afin que les deux cellules filles héritent de la même information génétique que la cellule mère. La double hélice de l'ADN fournit un mécanisme de réplication simple : les deux brins sont déroulés pour être séparés et chacun des deux brins sert de modèle pour recréer un brin à la séquence complémentaire par appariement entre bases nucléiques, ce qui permet de reconstituer deux hélices d'ADN bicaténaire identiques l'une à l'autre. Ce processus est catalysé par un ensemble d'enzymes parmi lesquelles les ADN polymérases sont celles qui complémentent les brins d'ADN déroulées pour reconstruire les deux brins complémentaires. Comme ces ADN polymérases ne peuvent polymériser l'ADN que dans le sens 5' vers 3', différents mécanismes interviennent pour copier les brins antiparallèles de la double hélice[74] :

Sur ce schéma, la Pol α devrait plutôt être représentée à droite de l'ADN ligase, à l'extrémité gauche du dernier fragment d'Okazaki formé sur le brin indirect.
Gènes et génome
[modifier | modifier le code]
L'ADN du génome est organisé et compacté selon un processus appelé condensation de l'ADN afin de pouvoir se loger dans l'espace restreint d'une cellule. Chez les eucaryotes, l'ADN est localisé essentiellement dans le noyau, avec une petite fraction également dans les mitochondries et, chez les plantes, dans les chloroplastes. Chez les procaryotes, l'ADN se trouve au sein d'une structure irrégulière du cytoplasme appelée nucléoïde[76]. L'information génétique du génome est organisée au sein de gènes, et l'ensemble complet de cette information est appelé génotype. Un gène est une fraction de l'ADN qui influence une caractéristique particulière de l'organisme et constitue de ce fait un élément de l'hérédité. Il contient un cadre de lecture ouvert qui peut être transcrit en ARN, ainsi que des séquences de régulation de l'expression génétique telles que les promoteurs et les amplificateurs qui en contrôlent la transcription.
Chez la plupart des espèces, seule une petite fraction du génome encode des protéines. Ainsi, environ 1,5 % du génome humain est constitué d'exons codant des protéines, tandis que plus de 50 % de l'ADN humain est constitué de séquences répétées non codantes[77] ; le reste de l'ADN code différents types d'ARN tels que des ARN de transfert et des ARN ribosomiques. La présence d'une telle quantité d'ADN non codant dans le génome des eucaryotes ainsi que la grande variabilité de la taille du génome des différents organismes — taille qui n'a aucun rapport avec la complexité des organismes correspondants — est une question connue depuis les débuts de la biologie moléculaire et souvent appelée paradoxe de la valeur C, cette « valeur C » désignant, chez les organismes diploïdes, la taille du génome, et un multiple de cette taille chez les polyploïdes[78]. Cependant, certaines séquences d'ADN qui n'encodent pas de protéines peuvent coder des molécules d'ARN fonctionnelles intervenant dans la régulation de l'expression génétique[79].
Certaines séquences d'ADN non codantes jouent un rôle structurel dans les chromosomes. Les télomères et les centromères contiennent généralement peu de gènes mais contribuent significativement aux fonctions biologiques et à la stabilité mécanique des chromosomes[39],[80]. Une fraction significative de l'ADN non codant est constituée de pseudogènes, qui sont des copies de gènes rendues inactives à la suite de mutations[81]. Ces séquences ne sont généralement que des fossiles moléculaires mais peuvent parfois servir de matière première génétique pour la création de nouveaux gènes à travers des processus de duplication génétique et de divergence évolutive[82].
Expression de l'information génétique
[modifier | modifier le code]
L'expression génétique consiste à convertir le génotype d'un organisme en phénotype, c'est-à-dire en un ensemble de caractéristiques propres à cet organisme. Ce processus est influencé par divers stimuli extérieurs et comprend les trois grandes étapes suivantes :
- transcription de l'ADN en ARN, un acide nucléique différent qui peut posséder une fonction biologique propre — ARN ribosomique, ARN de transfert, etc. — ou bien servir d'intermédiaire pour la biosynthèse des protéines — ARN messager ;
- traduction génétique de l'ARN messager en protéines ;
- activité physiologique des ARN non codants et des protéines au sein des organismes.
Notons qu'un même ADN peut à deux étapes du développement d'un organisme s'exprimer (en raison de répresseurs et dérépresseurs différents) de façons très dissemblables, l'exemple le plus connu étant celui de la chenille et du papillon, morphologiquement très éloignés.
Transcription
[modifier | modifier le code]
L'information génétique codée par la séquence des nucléotides des gènes de l'ADN peut être copiée sur un acide nucléique différent de l'ADN et appelé ARN. Cet ARN est structurellement très semblable à une molécule d'ADN monocaténaire mais en diffère par la nature de l'ose de ses nucléotides — l'ARN contient du ribose là où l'ADN contient du désoxyribose — ainsi que par l'une de ses bases nucléiques — la thymine de l'ADN y est remplacée par l'uracile.
La transcription de l'ADN en ARN est un processus complexe dont l'élucidation fut une avancée majeure de la biologie moléculaire au cours de la seconde moitié du XXe siècle. Elle est étroitement régulée, notamment par des protéines appelées facteurs de transcription qui, en réponse à des hormones par exemple, permettent la transcription de gènes cibles : c'est par exemple le cas des hormones sexuelles telles que les œstrogènes, la progestérone et la testostérone.
Traduction
[modifier | modifier le code]L'ARN issu de la transcription de l'ADN peut être non codant ou codant. Dans le premier cas, il possède une fonction physiologique propre dans la cellule ; dans le second cas, il s'agit d'un ARN messager, qui sert à transporter l'information génétique contenue dans l'ADN vers les ribosomes, qui organisent le décodage de cette information à l'aide de l'ARN de transfert. Ces ARN de transfert sont liés à un acide aminé parmi les 22 acides aminés protéinogènes et possèdent chacun un groupe de trois bases nucléiques consécutives appelées anticodon. Les trois bases de ces anticodons peuvent s'apparier à trois bases consécutives de l'ARN messager, ce triplet de bases formant un codon complémentaire de l'anticodon de l'ARN de transfert. La complémentarité du codon de l'ARN messager et de l'anticodon de l'ARN de transfert repose sur des règles d'appariement de type Watson-Crick régissant la structure secondaire des ADN bicaténaires.
Code génétique
[modifier | modifier le code]
La correspondance entre les 64 codons possibles et les 22 acides aminés protéinogènes est appelée code génétique. Ce code est matérialisé par les différents ARN de transfert réalisant physiquement la liaison entre un acide aminé donné et différents anticodons selon les différents ARN de transfert pouvant se lier à un même acide aminé. Ainsi, une séquence donnée de bases nucléiques au sein d'un gène sur l'ADN peut être convertie en une séquence précise d'acides aminés formant une protéine dans le cytoplasme de la cellule.
Il existe davantage de codons qu'il n'existe d'acides aminés à coder. On dit de ce fait que le code génétique est dégénéré. Outre les acides aminés protéinogènes, il code également la fin de traduction à l'aide de trois codons particuliers dits codons STOP : TAA, TGA et TAG sur l'ADN.
Interactions avec des protéines et des enzymes
[modifier | modifier le code]Toutes les fonctions biologiques de l'ADN dépendent d'interactions avec des protéines. Il peut s'agir d'interactions non spécifiques comme d'interactions avec des protéines qui se lient spécifiquement à une séquence précise de l'ADN. Des enzymes peuvent également se lier à l'ADN et, parmi celles-ci, les polymérases qui assurent la réplication de l'ADN ainsi que sa transcription en ARN jouent un rôle particulièrement déterminant.
Protéines
[modifier | modifier le code]

Protéines non spécifiques d'une séquence d'ADN
[modifier | modifier le code]Les protéines structurelles qui se lient à l'ADN offrent des exemples bien compris d'interactions non spécifiques entre des protéines et de l'ADN. Celui-ci est maintenu au sein des chromosomes en formant des complexes avec des protéines structurelles qui condensent l'ADN en une structure compacte appelée chromatine. Chez les eucaryotes, cette structure fait intervenir de petites protéines basiques appelées histones, tandis qu'elle fait intervenir de nombreuses protéines de différentes sortes chez les procaryotes[85],[86]. Les histones forment avec l'ADN un complexe en forme de disque appelé nucléosome contenant deux tours complets d'une molécule d'ADN bicaténaire enroulée autour de la protéine. Ces interactions non spécifiques s'établissent entre les résidus basiques des histones et le squelette acide constitué d'une alternance ose–phosphate portant les bases nucléiques de la double hélice d'ADN. Il se forme ainsi des liaisons ioniques indépendantes de la séquence des bases de l'ADN[87]. Ces résidus d'acides aminés basiques subissent des modifications chimiques telles que des méthylations, des phosphorylations et des acétylations[88]. Ces modifications chimiques modifient l'intensité des interactions entre l'ADN et les histones, rendant l'ADN plus ou moins accessible aux facteurs de transcription et modulant ainsi l'activité de transcription[89]. Parmi les autres protéines se liant à l'ADN de façon non spécifique, on compte les protéines nucléaires du groupe à haute mobilité électrophorétique, dites HMG, qui se lient à l'ADN courbé ou distordu[90]. Ces protéines sont importantes pour infléchir les réseaux de nucléosomes et les arranger en structures plus grandes qui constituent les chromosomes[91].
Parmi les protéines à interactions non spécifiques avec l'ADN, celles qui se lient spécifiquement à l'ADN monocaténaire constituent un groupe particulier. Chez l'homme, la protéine A en est le représentant le mieux compris. Elle intervient lorsque les deux brins d'une double hélice sont séparés, notamment lors de la réplication, la recombinaison et la réparation de l'ADN[92]. Ces protéines semblent stabiliser l'ADN monocaténaire et empêcher qu'il ne forme des structures en tige-boucle — épingle à cheveux — ou ne soit dégradé par des nucléases.
Protéines spécifiques d'une séquence d'ADN
[modifier | modifier le code]A contrario, d'autres protéines ne se lient qu'à des séquences d'ADN spécifiques. Parmi ces protéines, les plus étudiées sont les différents facteurs de transcription, qui sont des protéines régulant la transcription. Chaque facteur de transcription ne se lie qu'à un ensemble particulier de séquences d'ADN et active ou inhibe les gènes dont l'une de ces séquences spécifique est proche du promoteur. Les facteurs de transcription réalisent ceci de deux façons. Ils peuvent tout d'abord se lier à l'ARN polymérase responsable de la transcription, directement ou par l'intermédiaire d'autres protéines médiatrices ; ceci positionne la polymérase au niveau du promoteur et lui permet de commencer la transcription[93]. Ils peuvent également se lier à des enzymes qui modifient les histones au niveau du promoteur, ce qui a pour effet de modifier l'accessibilité de l'ADN pour la polymérase[94].
Dans la mesure où ces cibles d'ADN peuvent se distribuer dans tout le génome d'un organisme, un changement de l'activité d'un seul type de facteurs de transcription peut affecter des milliers de gènes[95]. Par conséquent, ces protéines sont souvent la cible de processus de transduction de signal contrôlant les réponses à des changements environnementaux, le développement ou la différenciation cellulaires. La spécificité de l'interaction de ces facteurs de transcription avec l'ADN vient du fait que ces protéines établissent de nombreux contacts avec les bords des bases nucléiques, ce qui leur permet de « lire » la séquence de l'ADN. La plupart de ces interactions ont lieu dans le grand sillon de la double hélice de l'ADN, là où les bases sont le plus accessibles[25].
Enzymes
[modifier | modifier le code]Nucléases
[modifier | modifier le code]
Les nucléases sont des enzymes qui clivent les brins d'ADN en catalysant l'hydrolyse des liaisons phosphodiester. Les nucléases qui clivent les nucléotides situés à l'extrémité des brins d'ADN sont appelées exonucléases, tandis que celles qui clivent les nucléotides situés à l'intérieur des brins d'ADN sont appelées endonucléases. Les nucléases les plus couramment utilisées en biologie moléculaire sont les enzymes de restriction, qui clivent l'ADN au niveau de séquences spécifiques. Ainsi l'enzyme EcoRV reconnaît la séquence de six bases 5′-GATATC-3′ et la clive en son milieu. In vivo, ces enzymes protègent les bactéries contre l'infection par des phages en digérant l'ADN de ces virus lorsqu'il pénètre dans la cellule bactérienne[97]. En ingénierie moléculaire, elles sont utilisées dans les techniques de clonage moléculaire et pour déterminer l'empreinte génétique.
ADN ligases
[modifier | modifier le code]
À l'inverse, les enzymes appelées ADN ligases peuvent recoller des brins d'ADN rompus ou clivés[98]. Ces enzymes sont particulièrement importantes au cours de la réplication de l'ADN car ce sont elles qui suturent les fragments d'Okazaki produits sur le brin retardé, appelé aussi brin indirect, au niveau de la fourche de réplication. Elles interviennent également dans les mécanismes de réparation de l'ADN et de recombinaison génétique[98].
ADN topoisomérases
[modifier | modifier le code]Les topoisomérases sont des enzymes ayant à la fois une activité nucléase et une activité ligase. L'ADN gyrase est un exemple de telles enzymes. Ces protéines modifient le taux de surenroulement de l'ADN en sectionnant une double hélice pour permettre aux deux segments formés de tourner l'un par rapport à l'autre en relâchant les supertours avant d'être à nouveau suturés l'un à l'autre[18]. D'autres types de topoisomérases sont capables de sectionner une double hélice pour permettre le passage d'un autre segment de double hélice à travers la brèche ainsi formée avant de refermer cette dernière[99]. Les topoisomérases sont indispensables à de nombreux processus impliquant l'ADN, tels que la transcription et la réplication de l'ADN[19].
Hélicases
[modifier | modifier le code]Les hélicases constituent des sortes de moteurs moléculaires. Elles utilisent l'énergie chimique de nucléosides triphosphate, essentiellement l'ATP, pour briser les liaisons hydrogène unissant les paires de bases et dérouler la double hélice d'ADN pour en libérer les deux brins[100]. Ces enzymes sont indispensables à la plupart des processus nécessitant que des enzymes accèdent aux bases de l'ADN.
ADN polymérases
[modifier | modifier le code]
Les ADN polymérases sont des enzymes qui synthétisent des chaînes de polynucléotides à partir de nucléosides triphosphates. La séquence des chaînes qu'elles synthétisent est déterminée par celle d'une chaîne de polynucléotides préexistante appelée matrice. Ces enzymes fonctionnent en ajoutant continuellement des nucléotides à l'hydroxyle de l'extrémité 3’ de la chaîne polypeptidique en cours de croissance. Pour cette raison, toutes les polymérases fonctionnent dans le sens 5’ vers 3’[101]. Le nucléoside triphosphate ayant une base complémentaire de celle de la matrice s'apparie à celle-ci dans le site actif de ces enzymes , ce qui permet aux polymérases de produire des brins d'ADN dont la séquence est exactement complémentaire de celle du brin matrice. Les polymérases sont classées en fonction du type de brins qu'elles utilisent.
Au cours de la réplication, les ADN polymérases ADN-dépendantes réalisent des copies de brins d'ADN. Afin de préserver l'information génétique, il est essentiel que la séquence des bases de chaque copie soit exactement complémentaire de la séquence des bases sur le brin matrice. Pour ce faire, de nombreuses ADN polymérases ont la capacité de corriger leurs éventuelles erreurs de réplication — fonction de proofreading. Elles sont pour cela capables d'identifier le défaut de formation d'une paire de bases entre le brin matrice et le brin en cours de croissance au niveau de la base qu'elles viennent d'insérer et de cliver ce nucléotide à l'aide d'une activité exonucléase 3’ → 5’ afin d'éliminer cette erreur de réplication[102]. Chez la plupart des organismes, les ADN polymérases fonctionnent au sein de grands complexes appelés réplisomes qui contiennent plusieurs sous-unités complémentaires telles que clamps — pinces à ADN — et hélicases[103].
Les ADN polymérases ARN-dépendantes sont une classe de polymérases spécialisées capables de copier une séquence d'ARN en ADN. Elles comprennent la transcriptase inverse, qui est une enzyme virale impliquée dans l'infection des cellules hôte par les rétrovirus, et la télomérase, enzyme indispensable à la réplication des télomères[38],[104]. La télomérase est une polymérase inhabituelle en ce qu'elle contient sa propre matrice d'ARN au sein de sa structure[39].
ARN polymérases
[modifier | modifier le code]La transcription est réalisée par une ARN polymérase ADN-dépendante qui copie une séquence d'ADN en ARN. Afin de commencer la transcription d'un gène, l'ARN polymérase se lie tout d'abord à une séquence de l'ADN appelée promoteur et sépare les brins d'ADN. Puis elle copie la séquence d'ADN constituant le gène en une séquence complémentaire d'ARN jusqu'à atteindre une région de l'ADN appelée terminateur, où elle s'arrête et se détache de l'ADN. Tout comme l'ADN polymérase ADN-dépendante, l'ARN polymérase II — enzyme qui transcrit la plupart des gènes du génome humain — fonctionne au sein d'un grand complexe protéique comprenant plusieurs sous-unités complémentaires et régulatrices[105].
Évolution de l'information génétique
[modifier | modifier le code]Mutations
[modifier | modifier le code]
Chaque division cellulaire est précédée d'une réplication de l'ADN conduisant à une réplication des chromosomes. Ce processus conserve normalement l'information génétique de la cellule, chacune des deux cellules filles héritant d'un patrimoine génétique complet identique à celui de la cellule mère. Il arrive cependant que ce processus ne se déroule pas normalement et que l'information génétique de la cellule s'en trouve modifiée. On parle dans ce cas de mutation génétique. Cette altération du génotype peut être sans conséquence ou, au contraire, altérer également le phénotype résultant de l'expression des gènes altérés.
Recombinaison génétique
[modifier | modifier le code]

Une double hélice d'ADN n'interagit généralement pas avec d'autres segments d'ADN et, dans les cellules humaines, les différents chromosomes occupent même chacun une région qui leur est propre au sein du noyau et appelée territoire chromosomique[107]. Cette séparation physique des différents chromosomes est essentielle au fonctionnement de l'ADN comme répertoire stable et pérenne de l'information génétique dans la mesure où l'une des rares fois où des chromosomes interagissent survient lors de l'enjambement responsable de la recombinaison génétique, c'est-à-dire lorsque deux doubles hélices d'ADN sont rompues, échangent leurs sections et se ressoudent.
La recombinaison permet aux chromosomes d'échanger du matériel génétique et de produire de nouvelles combinaisons de gènes, ce qui accroît l'efficacité de la sélection naturelle et peut être déterminant dans l'évolution rapide de nouvelles protéines[108]. La recombinaison génétique peut également survenir lors de la réparation de l'ADN, notamment en cas de rupture simultanée des deux brins de la double hélice d'ADN[109].
La forme la plus courante de recombinaison chromosomique est la recombinaison homologue, lors de laquelle les deux chromosomes en interaction partagent des séquences très semblables. Les recombinaisons non homologues peuvent fortement endommager les cellules car elles peuvent conduire à des translocations et des anomalies génétiques. La réaction de recombinaison est catalysée par des enzymes appelées recombinases, telle que la protéine Rad51[110]. La première étape de ce processus est une rupture des deux brins de la double hélice provoquée par une endonucléase ou par un dommage à l'ADN[111]. Une suite d'étapes catalysées par la recombinase conduit à réunir les deux hélices par au moins une jonction de Holliday dans laquelle un segment monocaténaire de chaque double hélice est ressoudé au brin complémentaire de l'autre double hélice. La jonction de Holliday est une jonction cruciforme qui, lorsque les brins ont des séquences symétriques, peut se déplacer le long de la paire de chromosomes en échangeant un brin avec l'autre. La réaction de recombinaison s'arrête par clivage de la jonction et suture de l'ADN libéré[112].
Éléments génétiques mobiles
[modifier | modifier le code]
L'information génétique codée par l'ADN n'est pas nécessairement fixe dans le temps et certaines séquences sont susceptibles de se déplacer d'une partie du génome à une autre. Ce sont les éléments génétiques mobiles. Ces éléments sont mutagènes et peuvent altérer le génome des cellules. On trouve parmi eux notamment les transposons et les rétrotransposons, ces derniers agissant, contrairement aux premiers, à travers un ARN intermédiaire redonnant une séquence d'ADN sous l'action d'une transcriptase inverse. Ils se déplacent au sein du génome sous l'effet de transposases, enzymes particulières qui les détachent d'un endroit et les recollent à un autre endroit du génome cellulaire, et seraient responsables de la migration de pas moins de 40 % du génome humain au cours de l'évolution d'Homo sapiens[113].
Ces éléments transposables constituent une fraction importante du génome des êtres vivants, notamment chez les plantes où ils représentent souvent l'essentiel de l'ADN nucléaire, comme chez le maïs où de 49 à 78 % du génome est constitué de rétrotransposons[114]. Chez le blé, près de 90 % du génome est formé de séquences répétées et 68 % d'éléments transposables[115]. Chez les mammifères, près de la moitié du génome — de 45 à 48 % — est constituée d'éléments transposables ou de rémanents de ces derniers, et environ de 42 % du génome humain est formé de rétrotransposons, tandis que 2 à 3 % est formé de transposons d'ADN[116]. Ce sont par conséquent des éléments importants du fonctionnement et de l'évolution du génome des organismes[117].
Les introns dits du groupe I et du groupe II sont d'autres éléments génétiques mobiles. Ce sont des ribozymes, c'est-à-dire de séquences d'ARN douées de propriétés catalytiques comme les enzymes, susceptibles d'autocatalyser leur propre épissage. Ceux du groupe I ont besoin de nucléotides à guanine pour fonctionner, contrairement à ceux du groupe II. Les introns du groupe I, par exemple, se retrouvent sporadiquement chez les bactéries, plus significativement chez les eucaryotes simples, et chez un très grand nombre de plantes supérieures. On les trouve enfin au sein de gènes d'un grand nombre de bactériophages de bactéries à Gram positif[118], mais de seulement quelques phages de bactéries à Gram négatif — par exemple le phage T4[118],[119],[120],[121].
Transfert horizontal de gènes
[modifier | modifier le code]L'information génétique d'une cellule peut évoluer sous l'effet de l'incorporation de matériel génétique exogène absorbé à travers la membrane plasmique. On parle de transfert horizontal de gènes, par opposition au transfert vertical découlant la reproduction des êtres vivants. Il s'agit d'un important facteur d'évolution chez de nombreux organismes[122], notamment chez les unicellulaires. Ce processus fait souvent intervenir des bactériophages ou des plasmides[123],[124].
Les bactéries en état de compétence sont susceptibles d'absorber directement une molécule d'ADN extérieure et de l'incorporer dans leur propre génome, processus appelé transformation génétique. Elles peuvent également obtenir cet ADN sous forme de plasmide d'une autre bactérie à travers le processus de conjugaison bactérienne. Enfin, elles peuvent recevoir cet ADN par l'intermédiaire d'un bactériophage (un virus) par transduction. Les eucaryotes peuvent également recevoir du matériel génétique exogène, à travers un processus appelé transfection.
Évolution
[modifier | modifier le code]
L'ADN recèle toute l'information génétique permettant aux êtres vivants de vivre, de croître et de se reproduire. On ignore cependant si, au cours des 4 milliards d'années de l'histoire de la vie sur Terre, l'ADN a toujours joué ce rôle. Une théorie propose que ce soit un autre acide nucléique, l'ARN, qui ait été le support de l'information génétique des premières formes de vies apparues sur notre planète[125],[126]. L'ARN aurait joué le rôle central dans une première forme de métabolisme cellulaire dans la mesure où il est susceptible à la fois de véhiculer de l'information génétique et de catalyser des réactions chimiques en formant des ribozymes[127]. Ce monde à ARN, dans lequel l'ARN aurait servi à la fois de support de l'hérédité et d'enzymes, aurait influencé l'évolution du code génétique à quatre bases nucléiques, lequel offre un compromis entre la précision du codage de l'information génétique favorisée par un nombre restreint de bases d'une part et l'efficacité catalytique des enzymes favorisée par un plus grand nombre de monomères d'autre part[128].
Il n'existe cependant aucune preuve directe de l'existence passée de systèmes métaboliques et génétiques différents de ceux que nous connaissons aujourd'hui dans la mesure où il demeure impossible d'extraire du matériel génétique de la plupart des fossiles. L'ADN ne persiste en effet plus d'un million d'années avant d'être dégradé en fragments courts. L'existence d'ADN intact plus ancien a été proposée, en particulier celui d'une bactérie viable extraite d'un cristal de sel vieux de 150 millions d'années[129], mais ces publications demeurent controversées[130],[131].
Certains constituants de l'ADN — l'adénine, la guanine et des composés organiques apparentés — peuvent avoir été formés dans l'espace[132],[133],[134]. Des constituants de l'ADN et de l'ARN tels que l'uracile, la cytosine et la thymine, ont également été obtenus en laboratoire dans des conditions reproduisant celles rencontrées dans le milieu interplanétaire et interstellaire à partir de composés plus simples tels que la pyrimidine, retrouvée dans des météorites. La pyrimidine, tout comme certains hydrocarbures aromatiques polycycliques (HAP) — les composés les plus riches en carbone détectés dans l'univers — pourraient se former dans les étoiles géantes rouges ou dans les nuages interstellaires[135].
Technologies de l'ADN
[modifier | modifier le code]Génie génétique
[modifier | modifier le code]
Des méthodes ont été développées permettant de purifier l'ADN des êtres vivants, telles que l'extraction au phénol-chloroforme, et le manipuler en laboratoire, telles que les enzymes de restriction et la PCR. La biologie et la biochimie modernes font un usage intensif de ces techniques au cours du clonage moléculaire (en). L'ADN recombinant est une séquence d'ADN synthétique assemblée à partir d'autres séquences d'ADN. De tels ADN peuvent transformer des organismes sous forme de plasmides ou à l'aide d'un vecteur viral[138]. Les organismes génétiquement modifiés (OGM) résultants peuvent être utilisés pour produire par exemple des protéines recombinantes, utilisées dans la recherche médicale[139], ou dans l'agriculture[140],[141].
Police scientifique et médecine légale
[modifier | modifier le code]
L'ADN extrait du sang, du sperme, de la salive, d'un fragment de peau ou d'un poil prélevé sur une scène de crime peut être utilisé en médecine légale pour déterminer l'empreinte génétique d'un suspect. À cette fin, la séquence de segments d'ADN tels que des séquences microsatellites ou des minisatellites est comparée avec celle d'individus choisis pour l'occasion ou déjà répertoriés dans des bases de données. Cette méthode est généralement d'une très grande fiabilité pour identifier l'ADN correspondant à celui d'un individu suspect[142]. L'identification peut cependant être rendue plus complexe si la scène de crime est contaminée par l'ADN de plusieurs personnes[143]. L'identification par empreinte génétique a été développée en 1984 par le généticien britannique Sir Alec Jeffreys[144] et a été utilisée pour la première fois en 1987 pour confondre un violeur et tueur en série[145].
Histoire et anthropologie
[modifier | modifier le code]Dans la mesure où l'ADN accumule des mutations au cours du temps qui sont transmises par hérédité, il recèle des informations historiques qui, lorsqu'elles sont analysées par des généticiens en comparant des séquences issues d'organismes aux histoires différentes, permettent de retracer l'histoire de l'évolution de ces organismes, c'est-à-dire leur phylogénie[146]. Cette discipline, mettant la génétique au service de la paléobiologie, offre un puissant outil d'investigation en biologie de l'évolution. En comparant des séquences d'ADN issues de différents individus d'une même espèce, les généticiens des populations peuvent étudier l'histoire de populations particulières d'êtres vivants, un domaine allant de la génétique écologique jusqu'à l'anthropologie. Ainsi, l'étude de l'ADN mitochondrial, de l'ADN-Y, puis de l'ADN autosomal au sein de différentes populations humaines est utilisée pour retracer les migrations d'Homo sapiens à travers la planète. L'haplogroupe X (ADNmt) a par exemple été étudié en paléodémographie afin d'évaluer la parenté éventuelle des Amérindiens d'Amérique du Nord-Est avec les populations ouest-européennes du Solutréen[147].
-
![(en) Arbre phylogénétique soulignant les trois domaines du vivant : les eucaryotes sont représentés en rouge, les archées en vert et les bactéries en bleu[148].](//upload.wikimedia.org/wikipedia/commons/thumb/7/78/Collapsed_tree_labels_simplified.png/500px-Collapsed_tree_labels_simplified.png) (en) Arbre phylogénétique soulignant les trois domaines du vivant : les eucaryotes sont représentés en rouge, les archées en vert et les bactéries en bleu[148].
(en) Arbre phylogénétique soulignant les trois domaines du vivant : les eucaryotes sont représentés en rouge, les archées en vert et les bactéries en bleu[148]. -
![Carte des migrations humaines déduite d'études phylogénétiques du génome mitochondrial humain[149].](//upload.wikimedia.org/wikipedia/commons/thumb/a/ab/World_map_of_prehistoric_human_migrations.jpg/462px-World_map_of_prehistoric_human_migrations.jpg)
![(en) Arbre phylogénétique soulignant les trois domaines du vivant : les eucaryotes sont représentés en rouge, les archées en vert et les bactéries en bleu[148].](/fr/Fichier:Collapsed_tree_labels_simplified.png)
![Carte des migrations humaines déduite d'études phylogénétiques du génome mitochondrial humain[149].](/fr/Fichier:World_map_of_prehistoric_human_migrations.jpg)
Bio-informatique
[modifier | modifier le code]
La bio-informatique fait intervenir la manipulation, la recherche et l'exploration de données biologiques, ce qui comprend les séquences d'ADN. Le développement de techniques de stockage et de recherche de séquences d'ADN ont conduit à des avancées informatiques largement utilisées par ailleurs, notamment en ce qui concerne les algorithmes de recherche de sous-chaînes, l'apprentissage automatique et la théorie des bases de données[150]. Les algorithmes de recherche de chaînes de caractères, qui permettent de trouver une suite de lettres incluse dans une suite de lettres plus longue, ont été développés pour rechercher des séquences spécifiques de nucléotides[151]. La séquence d'ADN peut être alignée avec d'autres séquences d'ADN afin d'identifier des séquences homologues et situer les mutations spécifiques qui les distinguent. Ces techniques, notamment l'alignement de séquences multiples, sont utilisées afin d'étudier les relations phylogénétiques et les fonctions des protéines[152].
Les répertoires de données représentant la séquence complète d'un génome, tels que ceux produits par le Projet génome humain, atteignent une taille telle qu'ils sont difficiles à utiliser sans les annotations qui identifient l'emplacement des gènes et des éléments de régulation sur chaque chromosome. Les régions des séquences d'ADN qui possèdent les motifs caractéristiques associés aux gènes codant des protéines ou des ARN fonctionnels peuvent être identifiés par des algorithmes de prédiction de gènes, qui permettent aux chercheurs de prédire la présence de produits géniques particuliers et leur fonction possible au sein d'un organisme avant même qu'ils soient isolés expérimentalement[153]. Des génomes entiers peuvent également être comparés, ce qui peut mettre en évidence l'histoire de l'évolution d'organismes particuliers et permettre d'étudier des événements complexes de cette évolution.
Nanotechnologies de l'ADN
[modifier | modifier le code]
Les nanotechnologies de l'ADN utilisent les propriétés uniques de reconnaissance moléculaire (en) de l'ADN et plus généralement des acides nucléiques afin de créer des complexes ramifiés d'ADN auto-assemblé doués de propriétés intéressantes[155]. De ce point de vue, l'ADN est utilisé comme matériau structurel plutôt que comme porteur d'une information biologique. Ceci a conduit à la création de réseaux périodiques bidimensionnels, qu'ils soient assemblés par briques ou par le procédé de l'origami d'ADN, ou tridimensionnels ayant une forme polyédrique[156]. On a également réalisé des nanomachines en ADN et des constructions par auto-assemblage algorithmique[157]. De telles structures en ADN ont pu être utilisées pour organiser l'arrangement d'autres molécules telles que des nanoparticules d'or et des molécules de streptavidine[158], une protéine qui forme des complexes très résistants avec la biotine. Les recherches en électronique moléculaire fondée sur l'ADN ont conduit la société Microsoft à développer un langage de programmation appelé DNA Strand Displacement[159] (DSD) utilisé dans certaines réalisations de composants nanoélectroniques moléculaires à base d'ADN[160],[161].
Stockage de données
[modifier | modifier le code]L'ADN étant utilisé par les êtres vivants pour stocker leur information génétique, certaines équipes de recherche l'étudient également comme support destiné au stockage d'informations numériques au même titre qu'une mémoire informatique. Les acides nucléiques présenteraient en effet l'avantage d'une densité de stockage de l'information considérablement supérieure à celle des médias traditionnels — théoriquement plus d'une dizaine d'ordres de grandeur — avec une durée de vie également très supérieure.
Il est théoriquement possible d'encoder jusqu'à deux bits de données par nucléotide, permettant une capacité de stockage atteignant 455 millions de téraoctets par gramme d'ADN monocaténaire demeurant lisibles pendant plusieurs millénaires y compris dans des conditions de stockage non idéales[162], et une technique d'encodage atteignant 215 000 téraoctets par gramme d'ADN a été proposée en 2017[163] ; à titre de comparaison, un DVD double face double couche contient à peine 17 gigaoctets pour une masse typique de 16 g — soit une capacité de stockage 400 milliards de fois moindre par unité de masse. Une équipe de l'Institut européen de bio-informatique est ainsi parvenue en 2012 à coder 757 051 octets sur 17 940 195 nucléotides[164], ce qui correspond à une densité de stockage d'environ 2 200 téraoctets par gramme d'ADN[165]. De son côté, une équipe suisse a publié en février 2015 une étude démontrant la robustesse de l'ADN encapsulé dans de la silice comme support durable de l'information[166].
Par ailleurs, d'autres équipes travaillent sur la possibilité de stocker des informations directement dans des cellules vivantes, afin par exemple d'encoder des compteurs sur l'ADN d'une cellule pour en déterminer le nombre de divisions ou de différenciations, ce qui pourrait trouver des applications dans les recherches sur le cancer et sur le vieillissement[167].
Histoire de la caractérisation de l'ADN
[modifier | modifier le code]




Découvertes de l'ADN et de sa fonction
[modifier | modifier le code]L'ADN a été isolé pour la première fois en 1869 par le biologiste suisse Friedrich Miescher sous la forme d'une substance riche en phosphore dans le pus de bandages chirurgicaux usagés. Comme cette substance se trouvait dans le noyau des cellules, Miescher l'appela nucléine[168],[169]. En 1878, le biochimiste allemand Albrecht Kossel isola le composant non protéique de cette « nucléine » — les acides nucléiques — puis en identifia les cinq bases nucléiques[170]. En 1919, le biologiste américain Phoebus Levene identifia les constituants des nucléotides, c'est-à-dire la présence d'une base, d'un ose et d'un groupe phosphate[171]. Il suggéra que l'ADN consistait en une chaîne de nucléotides unis les uns aux autres par leurs groupes phosphate. Il pensait que les chaînes étaient courtes et que les bases s'y succédaient de façon répétée selon un ordre fixe. En 1937, le physicien et biologiste moléculaire britannique William Astbury réalisa le premier diagramme de diffraction de l'ADN par cristallographie aux rayons X, montrant que l'ADN possède une structure ordonnée[172].
En 1927, le biologiste russe Nikolai Koltsov eut l'intuition que l'hérédité reposait sur une « molécule héréditaire géante » constituée de « deux brins miroirs l'un de l'autre qui se reproduiraient de manière semi-conservative en utilisant chaque brin comme modèle »[173]. Il considérait cependant que c'étaient des protéines qui portaient l'information génétique[174]. En 1928, le bactériologiste anglais Frederick Griffith réalisa une expérience célèbre qui porte dorénavant son nom et par laquelle il démontra que des bactéries vivantes non virulentes mises en contact avec des bactéries virulentes tuées par la chaleur pouvaient être transformées en bactéries virulentes[175],[176]. Cette expérience ouvrit la voie à l'identification en 1944 de l'ADN comme vecteur de l'information génétique à travers l'expérience d'Avery, MacLeod et McCarty[177]. Le biochimiste belge Jean Brachet démontra en 1946 que l'ADN est un constituant des chromosomes[178], et le rôle de l'ADN dans l'hérédité fut confirmé en 1952 par les expériences de Hershey et Chase qui démontrèrent que le matériel génétique du phage T2 est constitué d'ADN[179].
Découverte de la structure en double hélice
[modifier | modifier le code]La première structure en double hélice antiparallèle aujourd'hui reconnue comme modèle correct de l'ADN a été publiée en 1953 par le biochimiste américain James Watson et le biologiste britannique Francis Crick dans un article devenu classique de la revue Nature[2]. Ils travaillaient sur le sujet depuis 1951 au laboratoire Cavendish de l'université de Cambridge, et entretenaient à ce titre une correspondance privée avec le biochimiste autrichien Erwin Chargaff, à l'origine des règles de Chargaff, publiées au printemps 1952, selon lesquelles, au sein d'une molécule d'ADN, le taux de chacune des bases puriques est sensiblement égal au taux de l'une des deux bases pyrimidiques, plus précisément le taux de guanine est égal à celui de cytosine et que le taux d'adénine est égal à celui de thymine[180],[181], ce qui suggéra l'idée d'un appariement de l'adénine avec la thymine et de la guanine avec la cytosine.
En mai 1952, l'étudiant britannique Raymond Gosling, qui travaillait sous la direction de Rosalind Franklin dans l'équipe de John Randall, prit un cliché de diffraction aux rayons X (le cliché 51[182]) d'un cristal d'ADN fortement hydraté. Ce cliché fut partagé avec Crick et Watson à l'insu de Franklin et fut déterminant dans l'établissement de la structure correcte de l'ADN. Franklin avait par ailleurs indiqué aux deux chercheurs que l'ossature phosphorée de la structure devait être à l'extérieur de celle-ci, et non près de l'axe central comme on le pensait alors. Elle avait de surcroît identifié le groupe d'espace des cristaux d'ADN, qui permit à Crick de déterminer que les deux brins d'ADN sont antiparallèles[183]. Alors que Linus Pauling et Robert Corey publiaient un modèle moléculaire d'acide nucléique formé de trois chaînes entrelacées avec, conformément aux idées de l'époque, les groupes phosphate près de l'axe central et les bases nucléiques orientées vers l'extérieur[184], Crick et Watson finalisèrent en février 1953 leur modèle à deux chaînes antiparallèles ayant les groupes phosphate à l'extérieur et les bases nucléiques à l'intérieur de la double hélice, modèle aujourd'hui considéré comme la première structure correcte de l'ADN à avoir jamais été proposée.
Ces travaux furent publiés dans le numéro du 25 avril 1953 de la revue Nature à travers cinq articles décrivant la structure finalisée par Crick et Watson, ainsi que les preuves à l'appui de ce résultat. Dans le premier article, intitulé Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid, Crick et Watson indiquent : « il ne nous a pas échappé que l'appariement spécifique que nous avons postulé suggère immédiatement un mécanisme possible pour la réplication du matériel génétique »[2]. Cet article était suivi d'une publication du Britannique Maurice Wilkins et al. portant sur la diffraction de rayons X par de l'ADN B in vivo, ce qui appuyait l'existence de la structure en double hélice dans les cellules vivantes et pas seulement in vitro[185], et de la première publication des travaux de Franklin et Goslin sur les données qu'ils avaient obtenues par diffraction aux rayons X et leur propre méthode d'analyse[186].
Rosalind Franklin mourut en 1958 d'un cancer et ne reçut pas le prix Nobel de physiologie ou médecine décerné en 1962, « pour leurs découvertes relatives à la structure moléculaire des acides nucléiques et leur importance pour le transfert de l'information génétique dans la matière vivante », à Francis Crick, James Watson et Maurice Wilkins[187], qui n'eurent pas un mot pour créditer Franklin de ses travaux ; le fait qu'elle n'ait pas été associée à ce prix Nobel continue de faire débat[188].
Théorie fondamentale de la biologie moléculaire
[modifier | modifier le code]En 1957, Crick publia un document mettant en forme ce qui est aujourd'hui connu comme la théorie fondamentale de la biologie moléculaire en décrivant les relations entre l'ADN, l'ARN et les protéines, articulées autour de « l'hypothèse de l'adaptateur »[189]. La confirmation du mode de réplication semi-conservative de la double hélice est intervenue en 1958 avec l'expérience de Meselson et Stahl[190]. Crick et al. poursuivirent leurs travaux et montrèrent que le code génétique est fondé sur des triplets de bases nucléiques successifs appelés codons, ce qui permit le déchiffrement du code génétique lui-même par Robert W. Holley, Har Gobind Khorana et Marshall W. Nirenberg[191]. Ces découvertes marquèrent la naissance de la biologie moléculaire.
Arts
[modifier | modifier le code]La structure hélicoïdale de l'ADN a inspiré plusieurs artistes, le plus célèbre étant le peintre surréaliste Salvador Dalí, qui s'en inspire dans neuf tableaux entre 1956 et 1976, dont Paysage de papillon (Le Grand masturbateur dans un paysage surréaliste avec ADN)[192],[193] (1957-1958) et Galacidalacidesoxyribonucleicacid[194] (1963).
Notes et références
[modifier | modifier le code]Notes
[modifier | modifier le code]- Hormis certaines cellules très spécialisées telles que les érythrocytes, dépourvues de matériel génétique car dérivant de normoblastes par perte de leur noyau.
Références
[modifier | modifier le code]- Franck Marmoz ; Nicolas Chareyre ; Cédric Putanier, 600 questions de culture juridique générale, Paris, Ellipses, , 190 p. (ISBN 9 782340 067523), p. 113
- (en) J. D. Watson et F. H. C. Crick, « Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid », Nature, vol. 171, no 4356, , p. 737-738 (ISSN 0028-0836, PMID 13054692, DOI 10.1038/171737a0, Bibcode 1953Natur.171..737W, lire en ligne)
- (en) Marshal Mandelkern, John G. Elias, Don Eden et Donald M. Crothers, « The dimensions of DNA in solution », Journal of Molecular Biology, vol. 152, no 1, , p. 153-161 (PMID 7338906, DOI 10.1016/0022-2836(81)90099-1, lire en ligne)
- (en) S. G. Gregory, K. F. Barlow, K. E. McLay et al., « The DNA sequence and biological annotation of human chromosome 1 », Nature, vol. 441, no 7091, , p. 315-321 (PMID 16710414, DOI 10.1038/nature04727, Bibcode 2006Natur.441..315G, lire en ligne)
- (en) A. Ghosh et M. Bansal, « A glossary of DNA structures from A to Z », Acta Crystallographica Section D – Biological Crystallography, vol. 59, no 4, , p. 620-626 (PMID 12657780, DOI 10.1107/S0907444903003251, lire en ligne)
- (en) Peter Yakovchuk, Ekaterina Protozanova et Maxim D. Frank-Kamenetskii, « Base-stacking and base-pairing contributions into thermal stability of the DNA double helix », Nucleic Acids Research, vol. 34, no 2, janvier, p. 2006 (PMID 16449200, PMCID 1360284, DOI 10.1093/nar/gkj454, lire en ligne)
- (en) Saija Kiljunen, Kristo Hakala, Elise Pinta, Suvi Huttunen, Patrycja Pluta, Aneta Gador, Harri Lönnberg and Mikael Skurnik, « Yersiniophage ϕR1-37 is a tailed bacteriophage having a 270kb DNA genome with thymidine replaced by deoxyuridine », Microbiology, vol. 151, no 12, , p. 4093-4102 (PMID 16339954, DOI 10.1099/mic.0.28265-0, lire en ligne)
- (en) Jumpei Uchiyama, Iyo Takemura-Uchiyama, Yoshihiko Sakaguchi, Keiji Gamoh, Shin-ichiro Kato, Masanori Daibata, Takako Ujihara, Naoaki Misawa et Shigenobu Matsuzaki, « Intragenus generalized transduction in Staphylococcus spp. by a novel giant phage », The ISME Journal, vol. 8, no 9, , p. 1949-1952 (PMID 24599069, DOI 10.1038/ismej.2014.29, lire en ligne)
- (en) Marie Nguyen et Anne-Lise Haenni, « Expression strategies of ambisense viruses », Virus Research, vol. 93, no 2, , p. 141-150 (PMID 12782362, DOI 10.1016/S0168-1702(03)00094-7, lire en ligne)
- (en) Tetsuji Kakutani, Yuriko Hayano, Takaharu Hayashi et Yuzo Minobe, « Ambisense segment 3 of rice stripe virus: the first instance of a virus containing two ambisense segments », Journal of General Virology, vol. 72, no 2, , p. 465-468 (PMID 1993885, DOI 10.1099/0022-1317-72-2-465, lire en ligne)
- (en) Yafeng Zhu, Takahiko Hayakawa, Shigemitsu Toriyama et Mami Takahashi, « Complete nucleotide sequence of RNA 3 of rice stripe virus: an ambisense coding strategy », Journal of General Virology, vol. 72, no Part 4, , p. 763-767 (PMID 2016591, DOI 10.1099/0022-1317-72-4-763, lire en ligne)
- (en) Alexander Hüttenhofer, Peter Schattner, Norbert Polacek, « Non-coding RNAs: hope or hype? », Trends in Genetics, vol. 21, no 5, , p. 289-297 (PMID 15851066, DOI 10.1016/j.tig.2005.03.007, lire en ligne)
- (en) Stephen H. Munroe, « Diversity of antisense regulation in eukaryotes: Multiple mechanisms, emerging patterns », Journal of Cellular Biochemistry, vol. 93, no 4, , p. 664-671 (PMID 15389973, DOI 10.1002/jcb.20252, lire en ligne)
- (en) Izabela Makalowska, Chiao-Feng Lin et Wojciech Makalowski, « Overlapping genes in vertebrate genomes », Computational Biology and Chemistry, vol. 29, no 1, , p. 1-12 (PMID 15680581, DOI 10.1016/j.compbiolchem.2004.12.006, lire en ligne)
- (en) Zackary I. Johnson et Sallie W. Chisholm, « Properties of overlapping genes are conserved across microbial genomes », Genome Research, vol. 14, no 11, , p. 2268-2272 (PMID 15520290, PMCID 525685, DOI 10.1101/gr.2433104, lire en ligne)
- (en) Robert A. Lamb et Curt M. Horvath, « Diversity of coding strategies in influenza viruses », Trends in Genetics, vol. 7, no 8, , p. 261-266 (PMID 1771674, DOI 10.1016/0168-9525(91)90326-L, lire en ligne)
- (en) Craig J. Benham et Steven P. Mielke, « DNA Mechanics », Annual Review of Biomedical Engineering, vol. 7, , p. 21-53 (PMID 16004565, DOI 10.1146/annurev.bioeng.6.062403.132016, lire en ligne)
- (en) James J. Champoux, « DNA TOPOISOMERASES: Structure, Function, and Mechanism », Annual Review of Biochemistry, vol. 70, , p. 369-413 (PMID 11395412, DOI 10.1146/annurev.biochem.70.1.369, lire en ligne)
- (en) James C. Wang, « Cellular roles of DNA topoisomerases: a molecular perspective », Nature Reviews Molecular Cell Biology, vol. 3, no 6, , p. 430-440 (PMID 12042765, DOI 10.1038/nrm831, lire en ligne)
- (en) Hauke Clausen-Schaumann, Matthias Rief, Carolin Tolksdorf et Hermann E. Gaub, « Mechanical Stability of Single DNA Molecules », Biophysical Journal, vol. 78, no 4, , p. 1997-2007 (PMID 10733978, PMCID 1300792, DOI 10.1016/S0006-3495(00)76747-6, lire en ligne)
- (en) J. Isaksson, S. Acharya, J. Barman, P. Cheruku et J. Chattopadhyaya, « Single-stranded adenine-rich DNA and RNA retain structural characteristics of their respective double-stranded conformations and show directional differences in stacking pattern », Biochemistry, vol. 43, no 51, , p. 15996-16010 (PMID 15609994, DOI 10.1021/bi048221v, lire en ligne)
- (en) Tigran V. Chalikian, Jens Völker, G. Eric Plum et Kenneth J. Breslauer, « A more unified picture for the thermodynamics of nucleic acid duplex melting: A characterization by calorimetric and volumetric techniques », Proceedings of the National Academy of Sciences of the United States of America, vol. 96, no 14, , p. 7853-7858 (PMID 10393911, PMCID 22151, DOI 10.1073/pnas.96.14.7853, Bibcode 1999PNAS...96.7853C, lire en ligne)
- (en) Pieter L. DeHaseth et John D. Helmann, « Open complex formation by Escherichia coli RNA polymerase: the mechanism of polymerase-induced strand separation of double helical DNA », Molecular Microbiology, vol. 16, no 5, , p. 817-824 (PMID 7476180, DOI 10.1111/j.1365-2958.1995.tb02309.x, lire en ligne)
- (en) Maria Letizia Di Pietro, Giuseppina La Ganga, Francesco Nastasi et Fausto Puntoriero, « Ru(II)-Dppz Derivatives and Their Interactions with DNA: Thirty Years and Counting », Applied Sciences, vol. 11, no 7, , p. 3038 (ISSN 2076-3417, DOI 10.3390/app11073038, lire en ligne, consulté le )
- (en) C. O. Pabo et R. T. Sauer, « Protein-DNA Recognition », Annual Review of Biochemistry, vol. 53, , p. 293-321 (PMID 6236744, DOI 10.1146/annurev.bi.53.070184.001453, lire en ligne)
- (en) Hirak S. Basu, Burt G. Feuerstein, David A. Zarling, Richard H. Shaffer et Laurence J. Marton, « Recognition of Z-RNA and Z-DNA Determinants by Polyamines in Solution: Experimental and Theoretical Studies », Journal of Biomolecular Structure and Dynamics, vol. 6, no 2, , p. 299-309 (PMID 2482766, DOI 10.1080/07391102.1988.10507714, lire en ligne)
- (en) Timothy J. Richmond et Curt A. Davey, « The structure of DNA in the nucleosome core », Nature, vol. 423, no 6936, , p. 145-150 (PMID 12736678, DOI 10.1038/nature01595, Bibcode 2003Natur.423..145R, lire en ligne)
- (en) I. C. Baianu, « X-ray scattering by partially disordered membrane systems », Acta Crystallographica Section A. Crystal Physics, Diffraction, Theoretical and General Crystallography, vol. A34, no 5, , p. 751-753 (DOI 10.1107/S0567739478001540, Bibcode 1978AcCrA..34..751B, lire en ligne)
- (en) Nuri A. Temiz, Duncan E. Donohue, Albino Bacolla, Brian T. Luke, Jack R. Collins, « The Role of Methylation in the Intrinsic Dynamics of B- and Z-DNA », PloS One, vol. 7, no 4, , e35558 (PMID 22530050, PMCID 3328458, DOI 10.1371/journal.pone.0035558, lire en ligne)
- (en) Young-Min Kang, Jongchul Bang, Eun-Hae Lee, Hee-Chul Ahn, Yeo-Jin Seo, Kyeong Kyu Kim, Yang-Gyun Kim, Byong-Seok Choi et Joon-Hwa Lee, « NMR Spectroscopic Elucidation of the B−Z Transition of a DNA Double Helix Induced by the Zα Domain of Human ADAR1 », Journal of the American Chemical Society, vol. 131, no 32, , p. 11485-11491 (PMID 19637911, DOI 10.1021/ja902654u, lire en ligne)
- (en) Yeon-Mi Lee, Hee-Eun Kim, Chin-Ju Park, Ae-Ree Lee, Hee-Chul Ahn, Sung Jae Cho, Kwang-Ho Choi, Byong-Seok Choi et Joon-Hwa Lee, « NMR Study on the B–Z Junction Formation of DNA Duplexes Induced by Z-DNA Binding Domain of Human ADAR1 », Journal of the American Chemical Society, vol. 134, no 11, , p. 5276-5283 (PMID 22339354, DOI 10.1021/ja211581b, lire en ligne)
- (en) Richard R Sinden, DNA structure and function, Academic Press, , 398 p. (ISBN 0-12-645750-6)
- (en) Rich A, Norheim A, Wang AHJ, « The chemistry and biology of left-handed Z-DNA », Annual Review of Biochemistry, vol. 53, , p. 791–846 (PMID 6383204, DOI 10.1146/annurev.bi.53.070184.004043)
- (en) Ho PS, « The non-B-DNA structure of d(CA/TG)n does not differ from that of Z-DNA », Proc Natl Acad Sci USA, vol. 91, no 20, , p. 9549–9553 (PMID 7937803, PMCID 44850, DOI 10.1073/pnas.91.20.9549, Bibcode 1994PNAS...91.9549H)
- (en) Frédéric Pâques et James E. Haber, « Multiple Pathways of Recombination Induced by Double-Strand Breaks in Saccharomyces cerevisiae », Microbiology and Molecular Biology Reviews, vol. 63, no 2, , p. 349-404 (PMID 10357855, PMCID 98970, lire en ligne)
- (en) Irina Voineagu, Vidhya Narayanan, Kirill S. Lobachev et Sergei M. Mirkin, « Replication stalling at unstable inverted repeats: Interplay between DNA hairpins and fork stabilizing proteins », Proceedings of the National Academy of Sciences of the United States of America, vol. 105, no 29, , p. 9936-9941 (PMID 18632578, PMCID 2481305, DOI 10.1073/pnas.0804510105, lire en ligne)
- (en) Guy-Franck Richard, Alix Kerrest et Bernard Dujon, « Comparative Genomics and Molecular Dynamics of DNA Repeats in Eukaryotes », Microbiology and Molecular Biology Reviews, vol. 72, no 4, , p. 686–727 (PMID 19052325, PMCID 2593564, DOI 10.1128/MMBR.00011-08, lire en ligne)
- (en) Carol W. Greider et Elizabeth H. Blackburn, « Identification of a specific telomere terminal transferase activity in tetrahymena extracts », Cell, vol. 43, no 2, , p. 405-413 (PMID 3907856, DOI 10.1016/0092-8674(85)90170-9, lire en ligne)
- (en) Constance I. Nugent et Victoria Lundblad, « The telomerase reverse transcriptase: components and regulation », Genes & Development, vol. 12, no 8, , p. 1073-1085 (PMID 9553037, DOI 10.1101/gad.12.8.1073, lire en ligne)
- (en) Woodring E. Wright, Valerie M. Tesmer, Kenneth E. Huffman, Stephen D. Levene et Jerry W. Shay, « Normal human chromosomes have long G-rich telomeric overhangs at one end », Genes & Development, vol. 11, no 21, , p. 2801-2809 (PMID 9353250, PMCID 316649, DOI 10.1101/gad.11.21.2801, lire en ligne)
- (en) Sarah Burge, Gary N. Parkinson, Pascale Hazel, Alan K. Todd et Stephen Neidle, « Quadruplex DNA: sequence, topology and structure », Nucleic Acids Research, vol. 34, no 19, , p. 5402-5415 (PMID 17012276, PMCID 1636468, DOI 10.1093/nar/gkl655, lire en ligne)
- (en) Gary N. Parkinson, Michael P. H. Lee et Stephen Neidle, « Crystal structure of parallel quadruplexes from human telomeric DNA », Nature, vol. 417, no 6891, , p. 876-880 (PMID 12050675, DOI 10.1038/nature755, lire en ligne)
- (en) Nadrian C. Seeman, « DNA enables nanoscale control of the structure of matter », Quarterly Reviews of Biophysics, vol. 38, no 4, , p. 363-371 (PMID 16515737, PMCID 3478329, DOI 10.1017/S0033583505004087, lire en ligne)
- (en) Deshmukh N. Gopaul, Feng Guo et Gregory D. Van Duyne, « Structure of the Holliday junction intermediate in Cre–loxP site‐specific recombination », The EMBO Journal, vol. 17, no 14, , p. 4175-4187 (PMID 9670032, PMCID 1170750, DOI 10.1093/emboj/17.14.4175, lire en ligne)
- (en) Qidong Hu et Michael G. Rosenfeld, « Epigenetic regulation of human embryonic stem cells », Frontiers in Genetics, vol. 3, , p. 238 (PMID 23133442, PMCID 3488762, DOI 10.3389/fgene.2012.00238, lire en ligne)
- (en) Robert J. Klose et Adrian P. Bird, « Genomic DNA methylation: the mark and its mediators », Trends in Biochemical Sciences, vol. 31, no 2, , p. 89-97 (DOI 10.1016/j.tibs.2005.12.008, lire en ligne)
- (en) Adrian Bird, « DNA methylation patterns and epigenetic memory », Genes & Development, vol. 16, no 1, , p. 6-21 (PMID 11782440, DOI 10.1101/gad.947102, lire en ligne)
- (en) C. P. Walsh et G. L. Xu, « Cytosine Methylation and DNA Repair », Current Topics in Microbiology and Immunology, vol. 301, , p. 283-315 (PMID 16570853, DOI 10.1007/3-540-31390-7_11, lire en ligne)
- (en) Shulin Zhang, Barry W. Glickman et Johan G. de Boer, « Spontaneous mutation of the lacI transgene in rodents: Absence of species, strain, and insertion-site influence », Environmental and Molecular Mutagenesis, vol. 37, no 2, , p. 141–146 (ISSN 1098-2280, DOI 10.1002/em.1021, lire en ligne, consulté le )
- (en) David Ratel, Jean-Luc Ravanat, François Berger et Didier Wion, « N6-methyladenine: the other methylated base of DNA », BioEssays, vol. 28, no 3, , p. 309-315 (PMID 16479578, PMCID 2754416, DOI 10.1002/bies.20342, lire en ligne)
- (en) Eric Lieberman Greer, Mario Andres Blanco, Lei Gu, Erdem Sendinc, Jianzhao Liu, David Aristizábal-Corrales, Chih-Hung Hsu, L. Aravind, Chuan He et Yang Shi, « DNA Methylation on N6-Adenine in C. elegans », Cell, vol. 161, no 4, , p. 868-878 (PMID 25936839, DOI 10.1016/j.cell.2015.04.005, lire en ligne)
- (en) Ye Fu, Guan-Zheng Luo, Kai Chen, Xin Deng, Miao Yu, Dali Han, Ziyang Hao, Jianzhao Liu, Xingyu Lu, Louis C. Doré, Xiaocheng Weng, Quanjiang Ji, Laurens Mets et Chuan He, « N6-Methyldeoxyadenosine Marks Active Transcription Start Sites in Chlamydomonas », Cell, vol. 161, no 4, , p. 879-892 (PMID 25936837, DOI 10.1016/j.cell.2015.04.010, lire en ligne)
- (en) Guoqiang Zhang, Hua Huang, Di Liu, Ying Cheng, Xiaoling Liu, Wenxin Zhang, Ruichuan Yin, Dapeng Zhang, Peng Zhang, Jianzhao Liu, Chaoyi Li, Baodong Liu, Yuewan Luo, Yuanxiang Zhu, Ning Zhang, Shunmin He, Chuan He, Hailin Wang et Dahua Chen, « N6-Methyladenine DNA Modification in Drosophila », Cell, vol. 161, no 4, , p. 893-906 (PMID 25936838, DOI 10.1016/j.cell.2015.04.018, lire en ligne)
- (en) Skirmantas Kriaucionis et Nathaniel Heintz, « The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain », Science, vol. 324, no 5929, , p. 929-930 (PMID 19372393, PMCID 3263819, DOI 10.1126/science.1169786, lire en ligne)
- (en) Larry Simpson, « A base called J », Proceedings of the National Academy of Sciences of the United States of America, vol. 95, no 5, , p. 2037-2038 (PMID 9482833, PMCID 33841, DOI 10.1073/pnas.95.5.2037, Bibcode 1998PNAS...95.2037S, lire en ligne)
- (en) Janet H. Gommers-Ampt, Fred Van Leeuwen, Antonius L.J. de Beer, Johannes F.G. Vliegenthart, Miral Dizdaroglu, Jeffrey A. Kowalak, Pamela F. Crain et Piet Borst, « β-D-glucosyl-hydroxymethyluracil: A novel modified base present in the DNA of the parasitic protozoan T. brucei », Cell, vol. 75, no 6, , p. 1129–1136 (PMID 8261512, DOI 10.1016/0092-8674(93)90322-H, lire en ligne)
- (en) Henri G.A.M. van Luenen, Carol Farris, Sabrina Jan, Paul-Andre Genest, Pankaj Tripathi, Arno Velds, Ron M. Kerkhoven, Marja Nieuwland, Andrew Haydock, Gowthaman Ramasamy, Saara Vainio, Tatjana Heidebrecht, Anastassis Perrakis, Ludo Pagie, Bas van Steensel, Peter J. Myler et Piet Borst, « Glucosylated Hydroxymethyluracil, DNA Base J, Prevents Transcriptional Readthrough in Leishmania », Cell, vol. 150, no 5, , p. 909-921 (PMID 22939620, PMCID 3684241, DOI 10.1016/j.cell.2012.07.030, lire en ligne)
- (en) Dane Z. Hazelbaker et Stephen Buratowski, « Transcription: Base J Blocks the Way », Current Biology, vol. 22, no 22, , R960-R962 (PMID 23174300, PMCID 3648658, DOI 10.1016/j.cub.2012.10.010, lire en ligne)
- (en) Mike Cross, Rudo Kieft, Robert Sabatini, Matthias Wilm, Martin de Kort, Gijs A. van der Marel, Jacques H. van Boom, Fred van Leeuwen, Piet Borst, « The modified base J is the target for a nove lDNA‐binding protein in kinetoplastid protozoans », The EMBO Journal, vol. 18, no 22, , p. 6573-6581 (PMID 10562569, PMCID 1171720, DOI 10.1093/emboj/18.22.6573, lire en ligne)
- (en) Courtney DiPaolo, Rudo Kieft, Mike Cross et Robert Sabatini, « Regulation of Trypanosome DNA Glycosylation by a SWI2/SNF2-like Protein », Molecular Cell, vol. 17, no 3, , p. 441-451 (PMID 15694344, DOI 10.1016/j.molcel.2004.12.022, lire en ligne)
- (en) Saara Vainio, Paul-André Genest, Bas ter Riet, Henri van Luenen et Piet Borst, « Evidence that J-binding protein 2 is a thymidine hydroxylase catalyzing the first step in the biosynthesis of DNA base J », Molecular and Biochemical Parasitology, vol. 164, no 2, , p. 157-161 (PMID 19114062, DOI 10.1016/j.molbiopara.2008.12.001, lire en ligne)
- (en) Thierry Douki, Anne Reynaud-Angelin, Jean Cadet et Evelyne Sage, « Bipyrimidine Photoproducts Rather than Oxidative Lesions Are the Main Type of DNA Damage Involved in the Genotoxic Effect of Solar UVA Radiation », Biochemistry, vol. 42, no 30, , p. 9221-9226 (PMID 12885257, DOI 10.1021/bi034593c, lire en ligne)
- (en) Jean Cadet, Thierry Delatour, Thierry Douki, Didier Gasparutto, Jean-Pierre Pouget, Jean-Luc Ravanat, Sylvie Sauvaigo, « Hydroxyl radicals and DNA base damage », Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, vol. 424, nos 1-2, , p. 9-21 (PMID 10064846, DOI 10.1016/S0027-5107(99)00004-4, lire en ligne)
- (en) Kenneth B. Beckman et Bruce N. Ames, « Oxidative Decay of DNA », Journal of Biological Chemistry, vol. 272, no 32, , p. 19633-19636 (PMID 9289489, DOI 10.1074/jbc.272.32.19633, lire en ligne)
- (en) Kristoffer Valerie et Lawrence F Povirk, « Regulation and mechanisms of mammalian double-strand break repair », Oncogene, vol. 22, no 37, , p. 5792-5812 (PMID 12947387, DOI 10.1038/sj.onc.1206679, lire en ligne)
- (en) Jan H.J. Hoeijmakers, « DNA Damage, Aging, and Cancer », The New England Journal of Medicine, vol. 361, no 15, , p. 1475-1485 (PMID 19812404, DOI 10.1056/NEJMra0804615, lire en ligne)
- (en) Alex A. Freitas et João Pedro de Magalhães, « A review and appraisal of the DNA damage theory of ageing », Mutation Research/Reviews in Mutation Research, vol. 728, nos 1-2, , p. 12-22 (PMID 21600302, DOI 10.1016/j.mrrev.2011.05.001, lire en ligne)
- (en) Lynnette R. Ferguson et William A. Denny, « The genetic toxicology of acridines », Mutation Research/Reviews in Genetic Toxicology, vol. 258, no 2, , p. 123-160 (PMID 1881402, DOI 10.1016/0165-1110(91)90006-H, lire en ligne)
- (en) Trent D Stephens, Carolyn J.W Bunde et Bradley J Fillmore, « Mechanism of action in thalidomide teratogenesis », Biochemical Pharmacology, vol. 59, no 12, , p. 1489-1499 (PMID 10799645, DOI 10.1016/S0006-2952(99)00388-3, lire en ligne)
- (en) Alan M. Jeffrey, « DNA modification by chemical carcinogens », Pharmacology & Therapeutics, vol. 28, no 2, , p. 237-272 (PMID 3936066, DOI 10.1016/0163-7258(85)90013-0, lire en ligne)
- (en) M. F. Brana, M. Cacho, A. Gradillas, B. de Pascual-Teresa et A. Ramos, « Intercalators as Anticancer Drugs », Current Pharmaceutical Design, vol. 7, no 17, , p. 1745-1780 (PMID 11562309, DOI 10.2174/1381612013397113, lire en ligne)
- (en) J. Craig Venter, Mark D. Adams, Eugene W. Myers, Peter W. Li, Richard J. Mural, Granger G. Sutton et al., « The Sequence of the Human Genome », Science, vol. 291, no 5507, , p. 1304-1351 (PMID 11181995, DOI 10.1126/science.1058040, Bibcode 2001Sci...291.1304V, lire en ligne)
- (en) Mar Albà, « Replicative DNA polymerases », Genome Biology, vol. 2, no 1, , reviews3002.1–4 (PMID 11178285, PMCID 150442, DOI 10.1186/gb-2001-2-1-reviews3002, lire en ligne)
- (en) Y. Whitney Yin et Thomas A. Steitz, « Structural Basis for the Transition from Initiation to Elongation Transcription in T7 RNA Polymerase », Science, vol. 298, no 5597, , p. 1387-1395 (PMID