
 French
French Deutsch
DeutschNitrile – Wikipedia

Nitrile sind eine Gruppe chemischer Verbindungen mit der allgemeinen Formel R–C≡N. Die charakteristische funktionelle Gruppe der Nitrile ist das dreifach gebundene Stickstoffatom ≡N beziehungsweise die Nitril- oder Cyanogruppe –C≡N. Die Stoffgruppe leitet sich vom Cyanwasserstoff (Blausäure, H–C≡N) ab. Ein Beispiel hierfür ist das Acetonitril, H3C–C≡N, bei dem R eine Methylgruppe ist.
Nitrile sind polare Verbindungen und können eine Vielzahl von Reaktionen eingehen, weshalb sie eine große Bedeutung in der chemischen Synthese haben, auch im industriellen Bereich. Typische Reaktionen sind die Umsetzung zu Carbonsäuren (Hydrolyse) und die Reduktion zu Aminen oder Aldehyden. Über das Stickstoffatom können Nitrile an Metallatome koordinieren und so Komplexe bilden. Hergestellt werden Nitrile oft durch Dehydratisierung (Wasserabspaltung) aus Carbonsäureamiden und Oximen oder durch die Einführung einer Cyanogruppe mit Cyanidsalzen wie Kaliumcyanid.
Nitrile kommen in vielen Pflanzen und Tieren natürlich vor. Eine besonders wichtige Gruppe sind die cyanogenen Glycoside, die Blausäure freisetzen können und für die Giftigkeit vieler Pflanzen verantwortlich sind, darunter Aprikosenkerne und Bittermandeln. Bei Tieren kommen Nitrile inklusive cyanogener Glycoside vor allem bei Insekten und Tausendfüßern vor und dienen oft der Verteidigung. Nitrile kommen auch bei Bakterien, Pilzen und im Weltraum vor.
Nitrile sind wichtige Intermediate und Reagenzien für die Chemie und kommen als Zwischenstufe bei der Strecker-Synthese von Aminosäuren vor. Verbindungen wie Methylmethacrylat und Adiponitril werden großtechnisch aus Cyanwasserstoff hergestellt. Wichtige industrielle Produkte, bei denen es sich um Nitrile handelt, sind Acrylfasern, Sekundenkleber, die oft auf Cyanacrylaten basieren, und Nitrilkautschuk, der für medizinische Handschuhe verwendet wird. Acetonitril ist ein wichtiges Lösungsmittel. Einige Nitrile werden als Duftstoffe und Schädlingsbekämpfungsmittel eingesetzt. Durch ihre charakteristischen physikalischen und geometrischen Eigenschaften spielt die Nitrilgruppe außerdem eine wichtige Rolle beim zielgerichteten Design pharmazeutischer Wirkstoffe.
Inwieweit Cyanwasserstoff selbst den Nitrilen zuzurechnen ist, ist umstritten. Zumindest formal lässt es sich als Nitril der Ameisensäure auffassen, andererseits ist es ein Grenzfall, was die Einordnung von Verbindungen in die organische oder anorganische Chemie anbelangt und wird oft als anorganisch betrachtet, während die Stoffgruppe der Nitrile klar der organischen Chemie angehört.
Geschichte
[Bearbeiten | Quelltext bearbeiten]
Die ersten Synthesen von Nitrilen stammen aus dem 19. Jahrhundert. Bereits im Jahr 1815 stellte Joseph Louis Gay-Lussac Dicyan unter Verwendung von Silbercyanid her.[1] 1832 synthetisierten Friedrich Wöhler und Justus von Liebig gezielt Benzoylcyanid aus Benzoylchlorid. Weiterhin erhielten sie durch Dehydratisierung von Benzamid mit Bariumoxid einen „ölartigen Körper mit süßlich aromatischem Geruch“,[2] der erst später den Namen Benzonitril erhielt. Zwei Jahr später stellte Théophile-Jules Pelouze zum ersten Mal Propionitril her und bediente sich dabei einer klassischen Substitutionsreaktion. Die Prägung des Begriffs Nitrile für die Verbindungsgruppe wird Hermann Fehling zugeschrieben, um das Jahr 1844.[3] 1903 untersuchte Arthur Lapworth die Bildung von Cyanhydrinen durch Addition von Blausäure an Aldehyde und Ketone und entdeckte, dass das eigentliche Nucleophil das Cyanid-Ion ist, sodass Zusatz einer Base die Reaktionsgeschwindigkeit erhöht. Dabei handelte es sich um eine der ersten Untersuchungen eines organischen Reaktionsmechanismus.[4][5]
Lange Zeit waren Nitrile von primär akademischem Interesse. Zwischen dem Ersten und Zweiten Weltkrieg nahm der Umfang der Forschung aber deutlich zu.[3] Bis in die zweite Hälfte des 20. Jahrhunderts wurden mehrere großtechnische industrielle Prozesse entwickelt, bei denen Nitrile hergestellt oder verwendet werden. Ein wichtiges Beispiel ist die Entwicklung von Nylon (Polyamid 6.6) in den 1930er-Jahren, da Adiponitril ein Schlüssel-Intermediat bei dessen Herstellung ist, das selbst wiederum durch Hydrocyanierung von Butadien mit Cyanwasserstoff hergestellt wird.[6][7] Acrylnitril-Polymere sind etwa seit den 1920er-Jahren bekannt, erlangten aber erst gegen Ende der 1940er-Jahre eine größere Bedeutung in der Verwendung als synthetische Fasern.[8] Sekundenkleber auf Basis von Cyanacrylaten gibt es ebenfalls seit Ende der 1940er-Jahre.[9]
Nomenklatur
[Bearbeiten | Quelltext bearbeiten]
nach IUPAC: Butannitril (blau markiertes C-Atom zählt zur Hauptkette),
formal auch Propancarbonitril (blau markiertes C-Atom zählt zum Substituenten)
Die funktionelle Gruppe der Nitrile mit der C≡N-Dreifachbindung wird als Nitril- oder Cyanogruppe bezeichnet.[10] Ist das Nitril die funktionelle Gruppe höchster Rangordnung, so wird die Endung -nitril an den Namen der Ausgangsverbindung angehängt. Das dreifach gebundene Kohlenstoffatom wird, wie immer, für die Benennung des Grundgerüsts mitgezählt.[11] Alternativ kann (analog zu -carbonsäure) die Endung -carbonitril verwendet werden, dann wird das Kohlenstoffatom nicht zum Grundgerüst gezählt.[12] Diese Endung muss verwendet werden, wenn die Nitrilgruppe an einen Ring gebunden ist (wie beim Cyclopentancarbonitril[S 1]) oder wenn nicht alle Gruppen Teil des Grundgerüsts sind, was immer der Fall ist, wenn mehr als zwei Nitrilgruppen vorkommen, da diese nur am, Kettenende stehen können.[13] Entsprechend ihrer Verwandtschaft mit den Carbonsäuren (der Nitrilkohlenstoff hat die gleiche Oxidationsstufe wie der Carboxylkohlenstoff) werden Trivialnamen oft aus dem Namen der Carbonsäuren mit der Endung -onitril abgeleitet (zum Beispiel Benzoesäure zu Benzonitril).[14] Ist die Nitrilfunktion nicht von höchster Rangordnung im Molekül, so wird die Vorsilbe Cyan- mit entsprechender Positionsbezeichnung verwendet. Auch hier wird das dreifach gebundene Kohlenstoffatom nicht zum Grundgerüst gezählt.[13]
Abgrenzung zu verwandten Verbindungen
[Bearbeiten | Quelltext bearbeiten]
Nitrile sind isomer zu Isocyaniden (Isonitrilen). Diese tragen ebenfalls eine C≡N-Dreifachbindung, allerdings ist der zugehörige Rest über das Stickstoffatom gebunden, weshalb es sich um Zwitterionen handelt.[15]
Verbindungen, bei denen an das Kohlenstoffatom einer C≡N-Gruppe ein Sauerstoffatom anschließt, werden als Cyanate bezeichnet.[16] Handelt es sich statt dem Sauerstoffatom um ein Schwefel- oder Selenatom, spricht man von Thiocyanaten und Selenocyanaten.[17][18] Ist die Cyanogruppe an ein Stickstoffatom gebunden, spricht man von einem Cyanamid.[19]
Neben den Nitrilen sind weitere Verbindungsklassen bekannt, die über C≡N-Dreifachbindung verfügen, wobei das Stickstoffatom eine vierte Bindung trägt, sodass es positiv geladen ist. Bei den Nitriloxiden ist zusätzlich ein Sauerstoffatom an das Stickstoffatom gebunden.[20] Handelt es sich stattdessen um ein Schwefel- oder um ein weiteres Stickstoffatom, spricht man von einem Nitrilsulfid oder einem Nitrilimid.[21][22] Ist das Stickstoffatom des Nitrils protoniert oder trägt einen weiteren organischen Rest, handelt es sich um ein Nitriliumion.[23] Trägt das Stickstoffatom einen organischen Rest mit einem negativ geladenen Kohlenstoffatom, handelt es sich um ein Nitril-Ylid, eine Form von Yliden.[24]
Vertreter und Eigenschaften
[Bearbeiten | Quelltext bearbeiten]Beide Atome der Nitrilgruppe sind sp-hybridisiert. Die Struktur der Nitrilgruppe ähnelt der C≡C-Dreifachbindung in Ethinylgruppen.[14] Die C≡N-Dreifachbindung ist im Vergleich zu anderen Verbindungen kurz. Im Acetonitril und Propionitril beträgt die Bindungslänge beispielsweise jeweils knapp 116 pm während die daran anschließende C-C-Einfachbindung jeweils etwa 147 pm lang ist.[25] Durch die Elektronegativitätsdifferenz zwischen Kohlenstoff und Stickstoff weisen Nitrile ein erhebliches Dipolmoment auf. Aus dem gleichen Grund liegen ihre Siedepunkte etwa 60 °C höher als die von Alkinen analoger Struktur oder vergleichbarer Molmasse.[26] Nitrile sind allgemein gute Lösungsmittel sowohl für polare als auch für unpolare Stoffe. Acetonitril und Propionitril sind mit Wasser mischbar, bei den längeren aliphatischen Nitrilen nimmt die Wasserlöslichkeit aber deutlich ab.[27]
Acidität
[Bearbeiten | Quelltext bearbeiten]Durch die stark elektronenziehenden Eigenschaften der Nitrilgruppe weisen die Verbindungen eine relevante CH-Acidität auf und können in α-Position (am Kohlenstoffatom neben der Nitrilgruppe) vergleichsweise leicht deprotoniert werden.[28] Neben der Elektronegativität des Stickstoffs spielt auch die Delokalisierung von Elektronen durch Mesomerie eine Rolle. So kann für ein α-deprotoniertes Nitril eine Grenzformel formuliert werden, bei der die negative Ladung auf dem Stickstoff liegt.[29] Malonitril mit zwei Nitrilgruppen hat einen pKs-Wert von etwa 11.[30] Cyanoform[S 2] mit drei Nitrilgruppen gehört mit einem pKs-Wert von −5,1 zu den stärksten organischen Säuren überhaupt[31] und ist saurer als Schwefelsäure (pKs-Wert −3,0) oder Salpetersäure (pKs-Wert −1,5).[32] Weitere Beispiele für Nitrile mit sehr niedrigem pKs-Wert sind die Cyanoderivate von Cyclopentadien, so hat Pentacyanocyclopentadien einen geschätzten pKs-Wert von −11.[33]
Spektroskopische Eigenschaften
[Bearbeiten | Quelltext bearbeiten]In der IR-Spektroskopie tritt durch die Streckschwingung der C≡N-Dreifachbindung eine deutliche Bande bei einer Wellenzahl von etwa 2250 cm−1 auf. Ist die Nitrilgruppe mit einer Alkendoppelbindung konjugiert, tritt die Bande bei etwa 2225 cm−1 auf. Ist die Nitrilgruppe an einen aromatischen Ring gebunden, liegt die Bande meist zwischen den beiden Werten.[34] Im 1H-NMR treten Protonen, die α zur Nitrilgruppe stehen bei etwa 2 ppm (1,98 ppm in Acetonitril) auf, bei einer ähnlichen Verschiebung wie Protonen, die α zu anderen Carbonsäurederivaten stehen (Carbonsäure oder Carbonsäureamid). Das Kohlenstoffatom der Nitrilfunktion tritt im 13C-NMR bei etwa 112–126 ppm auf.[14]
Vertreter
[Bearbeiten | Quelltext bearbeiten]Eine Auswahl einfacher Nitrile ist in der folgenden Tabelle mit Schmelz- und Siedepunkten angegeben. Das einfachste Nitril ist Acetonitril. Die Alkannitrile mit bis zu vierzehn Kohlenstoffatomen, das heißt bis einschließlich Tetradecannitril,[S 3] sind alle bei Raumtemperatur flüssig.[27] Das Gleiche gilt auch für das einfachste α,β-ungesättigte Nitril, Acrylnitril und das einfachste aromatische Nitril, Benzonitril. Die einfachsten Dinitrile sind Dicyan und Malonitril. Acetoncyanhydrin ist ein wichtiger Vertreter der Cyanhydrine. Im Vergleich dazu ist Cyanwasserstoff leicht flüchtig, da sein Siedepunkt etwa bei Raumtemperatur (26 °C) liegt.[35]
| Summenformel | Molmasse (g/mol) | Name | Strukturformel | CAS-Nummer | Schmelzpunkt (°C) | Siedepunkt (°C) | Referenz |
|---|---|---|---|---|---|---|---|
| C2H3N | 41,05 | Acetonitril | 75-05-8 | −45 | 82 | [36] | |
| C3H5N | 55,08 | Propionitril | 107-12-0 | −103 | 97 | [37] | |
| C4H7N | 69,11 | Butyronitril |  | 109-74-0 | −112 | 117 | [38] |
| C4H7N | 69,11 | Isobutyronitril |  | 78-82-0 | −72 | 104 | [39] |
| C3H3N | 53,06 | Acrylnitril |  | 107-13-1 | −82 | 77 | [40] |
| C7H5N | 103,12 | Benzonitril |  | 100-47-0 | −13 | 191 | [41] |
| C2N2 | 52,04 | Dicyan | 460-19-5 | −27,83 | −21,15 | [42] | |
| C3H2N2 | 66,06 | Malonitril |  | 109-77-3 | 32–34 | 220 | [43] |
| C4H7NO | 85,11 | Acetoncyanhydrin |  | 75-86-5 | −20 | 82 | [44] |
Toxikologie
[Bearbeiten | Quelltext bearbeiten]Die Aufnahme von Cyanwasserstoff führt selbst bei kleinen Mengen leicht zur Cyanidvergiftung.[45] Für die Toxizität vieler Nitrile spielt die Freisetzung von Cyanwasserstoff eine Rolle, sie ist jedoch nicht allein dafür verantwortlich.[27][46] Aliphatische Nitrile können beim Einatmen über die Lunge aufgenommen werden und sie durchdringen Haut und Schleimhäute.[27] Malonitril setzt im Metabolismus besonders viel Cyanwasserstoff frei, Acetonitril besonders wenig. Malonitril kann außerdem, ebenso wie das Cyanidion, die Cytochrom-c-Oxidase hemmen. Bei ungesättigten Nitrilen wie Allylcyanid und Acrylnitril steht eine cholinerge Symptomatik im Vordergrund. Während bei ihnen die Freisetzung von Cyanwasserstoff nur eine untergeordnete Rolle spielt, vermögen sie mit Thiolgruppen, wie sie in Proteinen des Körpergewebes vorkommen, zu reagieren und dadurch gesundheitsschädliche Auswirkungen zu zeitigen.[46] Viele Nitrile verursachen neurologische Symptome, die vermutlich auf die Freisetzung von Cyanwasserstoff zurückzuführen sind. Weitere Symptome, die vermutlich nicht mit der HCN-Freisetzung zusammenhängen, sind Reizungen von Atemwegen und Verdauungstrakt, Übelkeit, sowie Leber- und Nierenschäden.[27]
Während die Freisetzung von Cyanwasserstoff bei den meisten Nitrilen von untergeordneter Bedeutung ist, basiert die Giftigkeit der cyanogenen Glycoside direkt auf der Bildung von Cyanwasserstoff beziehungsweise Cyanid. Diese sind auch für die mögliche Giftigkeit verschiedener Speisepflanzen verantwortlich, darunter Maniok, Aprikosenkerne und Leinsamen.[47] Cyanid als Anion des Cyanwasserstoffs bindet in den Mitochondrien an Eisen-Cofactoren der Cytochrom-c-Oxidase, was den Sauerstoff-Umsatz und die ATP-Produktion in den Zellen verhindert, unabhängig von der Verfügbarkeit von Sauerstoff. Dies führt zunächst zu Symptomen wie erhöhtem Blutdruck, Hyperventilation, Herzrasen und Kopfschmerzen und schließlich zu Stupor, Krämpfen und Atemstillstand.[45]
Vorkommen
[Bearbeiten | Quelltext bearbeiten]Schon in den 90er-Jahren waren über 100 natürlich vorkommende Nitrile bekannt,[48] inzwischen sind mehrere hundert bekannt.[49] Die Verbindungen kommen in Bakterien, Pilzen, Pflanzen, sowie Gliederfüßern und Schwämmen vor.[48][49] Die Biosynthese natürlich vorkommender Nitrile geht oft von Aminosäuren aus. Deren N-Hydroxylierung und Decarboxylierung (Abspaltung der Carbonsäuregruppe als Kohlenstoffdioxid) ergibt Aldoxime, die die direkten Vorläufer der Nitrile sind.[48]
Vorkommen in Pflanzen
[Bearbeiten | Quelltext bearbeiten]Viele Nitrile kommen als Sekundärmetaboliten in Pflanzen vor.
Im Wunderbaum (Ricinus communis) kommt neben dem hochgiftigen Protein Ricin auch das Alkaloid Ricinin vor, das eine Nitrilfunktion trägt.[50] Das strukturell eng verwandte Nudiflorin[S 4] kommt in Trevia nudiflora (Familie Wolfsmilchgewächse) vor.[51] In braunem Senf kommt Indolacetonitril vor, das aus Indolacetaldoxim gebildet wird und vermutlich zur Verteidigung gegen pathogene Pilze dient.[52] In Jojoba kommt neben anderen Nitrilen Simmondsin vor, ein Glycosid mit einem α,β-ungesättigten Nitril im Aglycon.[53] Eine ähnliche Verbindung, das Menisdaurin, kommt in der Gewöhnlichen Stechpalme (Ilex aquifolium) vor.[54] Auch in mehreren Arten der Gattung Acacia kommen α,β-ungesättigte Nitrile vor, darunter das Sutherlandin[S 5] und das Acacipetalin.[55][56][S 6] Im Meerrettichbaum (Moringa oleifera) kommt Niazirin[S 7] vor, ein Glycosid von 4-Hydroxyphenylacetonitril.[57] Die Duftende Platterbse (Lathyrus odoratus) verursacht die Krankheit Lathyrismus, wofür N-Glutamyl-3-aminopropionitril[S 8] und sein Abbauprodukt 3-Aminopropionitril verantwortlich sind.[58][59] Das ätherische Öl von Heracleum transcaucasicum (Gattung Bärenklau) enthält Geranylnitril.[60] Pyridin-3-carbonitril kommt im Einjährigen Bingelkraut vor.[61] Cyanolipide sind eine Klasse von Lipiden, die ausschließlich in Seifenbaumgewächsen (Sapindaceae) vorkommen. Die Alkoholkomponente ist bei diesen ein ungesättigtes Nitril mit fünf Kohlenstoffatomen und ein oder zwei Hydroxygruppen, im Gegensatz zum Glycerin in den Glyceriden. Zu den Seifenbaumgewächsen, die Cyanolipide enthalten, gehören Waschnussbaum und Guaraná.[62][63] Cyanwasserstoff wird von vielen Pflanzen freigesetzt, die entsprechende cyanogene Verbindungen enthalten, vor allem cyanogene Glycoside und Cyanolipide.[64] In Pflanzen wirkt Cyanwasserstoff auch als Signalmolekül.[65]
-
 Ricinuspflanze
Ricinuspflanze -
 Ricinin
Ricinin -
 Guaraná, ein Seifenbaumgewächs, enthält Cyanolipide
Guaraná, ein Seifenbaumgewächs, enthält Cyanolipide -
 Eine Alkoholkomponente von Cyanolipiden, die zum Beispiel in Guaraná vorkommt
Eine Alkoholkomponente von Cyanolipiden, die zum Beispiel in Guaraná vorkommt


_fruits_(29055398276).jpg)

Nitrile aus Glucosinolaten in Kreuzblütlern
[Bearbeiten | Quelltext bearbeiten]Eine Gruppe von Naturstoffen, die als Vorläufer von Nitrilen auftreten, sind die Glucosinolate (Senfölglycoside), die ähnlich wie bei der direkten Biosynthese von Nitrilen über ein Aldoxim gebildet werden.[48] Die Glucosinolate sind eine wichtige Gruppe von Sekundärmetaboliten, die Pflanzen der Familie der Kreuzblütler (Brassicaceae) zur Verteidigung gegen Fressfeinde und Mikroorganismen dienen. Glucosinolate werden durch Myrosinase im Normalfall zu Isothiocyanaten abgebaut; in Gegenwart eines zusätzlichen Proteins (epithio specifier protein) jedoch unter anderem zu Nitrilen.[66][67] Sinigrin kommt vor allem in Meerrettich, Wasabi und braunem Senf vor, aber auch in Kopfkohl, Grünkohl, Blumenkohl und Rosenkohl, und wird neben Allylisothiocyanat unter anderem zu Allylcyanid (3-Butennitril) abgebaut.[68][69] Glucotropaeolin, das in Gartenkresse vorkommt, wird unter anderem zu Phenylacetonitril abgebaut, Gluconasturtiin, das in Brunnenkresse vorkommt, unter anderem zu Phenylpropionitril.[70] Sinalbin, das in Pfeilkresse vorkommt, kann analog zu 4-Hydroxyphenylacetonitril abgebaut werden.[71]
-
 Brunnenkresse
Brunnenkresse -
 Struktur des Gluconasturtiins
Struktur des Gluconasturtiins -
 Struktur des Phenylpropionitrils
Struktur des Phenylpropionitrils



Cyanhydrine und cyanogene Glycoside
[Bearbeiten | Quelltext bearbeiten]Cyanhydrine und deren Glycoside, die als cyanogene Glycoside bezeichnet werden, sind in der Natur weitverbreitet und kommen in mehreren Tausend Pflanzenarten vor.[5][49] Über einhundert natürlich vorkommende cyanogene Glycoside sind bekannt.[49] Cyanogene Glycoside dienen Pflanzen zur Verteidigung, aber möglicherweise auch als Speicherform für Stickstoff. Sie werden ausgehend von einer kleinen Zahl Aminosäuren und diversen Zuckern gebildet.[5] Bei Beschädigung der Pflanze kommen die Glycoside mit Enzymen (β-Glucosidasen und α-Hydroxynitrillyasen) in Kontakt, die zuerst das Aglycon (ein Cyanhydrin) freisetzen und dieses dann zu einer Carbonylverbindung und giftiger Blausäure abbauen. Amygdalin ist ein Glycosid des Mandelonitrils und eines der am weitesten verbreiteten cyanogenen Glycoside und kommt insbesondere in den Samen der Rosengewächse (Rosaceae) vor, darunter Apfel, Aprikose, Pfirsich, Pflaume, Kirsche und Mandel.[72] Während Amygdalin nur in Kernen von Pfirsichen vorkommt, enthalten die sonstigen Teile der Pflanze vorwiegend Prunasin.[73] Prunasin ist ebenfalls ein Glycosid des Mandelonitrils, allerdings ist die Zuckereinheit ein Monosaccharid (kein Disaccharid wie bei Amygdalin). Prunasin kommt als biosynthetischer Vorläufer von Amygdalin in Mandeln und Bittermandeln vor.[74] Prunasin kommt außerdem in Kirschlorbeer vor.[75] In Passionsfrüchten kommen Prunasin und Sambunigrin, sowie einige andere cyanogene Glycoside vor; in Papaya hauptsächlich Prunasin.[76][77] Sambunigrin, ebenfalls ein Glycosid von Mandelonitril, kommt auch in mehreren Arten der Gattung Holunder (Sambucus) vor, unter anderem im Schwarzen Holunder und im Kanadischen Holunder,[78][79] sowie in Ximenia americana.[80] Vicianin, ein weiteres Glycosid des Mandelonitrils, kommt in Farnen der Gattung Davellia (Familie Davelliaceae) vor.[81] Dhurrin ist ein cyanogenes Glycosid des 4-Hydroxymandelonitrils,[S 9] das in Sorghumhirse und anderen Arten der Gattung Sorghum vorkommt, darunter in Sorghum halepense.[82][83] Linamarin (mit dem Aglycon Acetoncyanhydrin) und Lotaustralin (mit dem Agylcon Butanoncyanhydrin) treten in den Gattungen Linum (zum Beispiel in Flachs) und Lotus sowie in der Gartenbohne gemeinsam auf.[84] Auch in Maniok kommen beide Verbindungen vor.[85] In der Mistelart Loranthus micranthus (Gattung Loranthus) kommt Linamaringallat vor, ein Derivat bei dem Linamarin zusätzlich mit Gallussäure verestert ist.[86] Der Kautschukbaum enthält ebenfalls Linamarin und Studien haben ergeben, dass die Verbindung in diesem Fall wahrscheinlich auch eine wichtige Speichersubstanz ist und nicht nur der Verteidigung dient. Die Samen enthalten besonders große Mengen der Verbindung, die bei der Entwicklung der Keimlinge umgewandelt wird, ohne dass Blausäure freigesetzt wird, was darauf hindeutet, dass das Linamarin für andere biosynthetische Prozesse verwendet wird.[87]
-
 Pfirsichbaum
Pfirsichbaum -
 Struktur des Amygdalins
Struktur des Amygdalins -
 Struktur des Prunasins
Struktur des Prunasins -
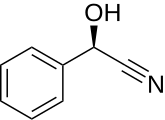 Mandelonitril, das Aglycon von Amygdalin und Prunasin
Mandelonitril, das Aglycon von Amygdalin und Prunasin



-mandelonitrile-2D-skeletal.svg)
Vorkommen in Tieren
[Bearbeiten | Quelltext bearbeiten]In vielen Gliederfüßern (Arthropoda) kommen cyanogene (Blausäure freisetzende) Nitrilverbindungen vor, unter anderem in Hundertfüßern (Chilopoda), Doppelfüßern (Diplopoda), Schnabelkerfe (Hemiptera), Käfern (Coleoptera) und Schmetterlingen (Lepidoptera).[88] Im Stachelbeerspanner (Abraxas grossulariata) kommt ein nitrilhaltiges Glycosid, Sarmentosin, vor, das vermutlich der Verteidigung dient.[89] Sarmentosin kommt außerdem in mehreren Arten der Gattung Parnassius vor.[90] Mehrere Arten der Glasflügelwanzen (unter anderem Jadera haematoloma) enthalten Cyanolipide beziehungsweise Cardiospermin,[S 10] die sie möglicherweise aus ihren Nahrungspflanzen sequestrieren, das heißt aufnehmen und einlagern.[88][91][92] Sechsfleck-Widderchen sind Schmetterlinge, die die cyanogenen Glycoside Linamarin und Lotaustralin sowohl aus ihren Nahrungspflanzen sequestrieren als auch selbst synthetisieren können.[93] Auch andere Arten aus der gleichen Gattung (Zygaena) wie das Sumpfhornklee-Widderchen enthalten cyanogene Glycoside.[94] Das Wehrsekret des Hundertfüßers Himantarium gabrielis enthält Benzoylcyanid, Phenylacetonitril, Mandelonitril (Benzaldehydcyanhydrin) und Mandelonitrilbenzoat.[95] Phenylacetonitril kommt auch als Hormon bei der Wüstenheuschrecke (Schistocerca gregaria) vor.[96] Bei verschiedenen Bandfüßern (Polydesmida) enthält das Wehrsekret ebenfalls Benzoylcyanid.[97] In der Milbenart Oribatula tibialis (Ordnung Hornmilben, Oribatida) kommt Mandelonitrilhexanoat vor.[98] Als Abbauprodukt der cyanogenen Verbindungen kommt in Gliederfüßern auch Cyanwasserstoff vor.[65]
-
 Stachelbeerspanner
Stachelbeerspanner -
 Wüstenheuschrecke
Wüstenheuschrecke -
 Himantarium gabrielis
Himantarium gabrielis -
 Benzoylcyanid kommt in den Wehrsekreten verschiedener Arthropoda vor
Benzoylcyanid kommt in den Wehrsekreten verschiedener Arthropoda vor
_The_Magpie_(Abraxas_grossulariata)_(14502341918).jpg)



Neben den Arthropoda enthalten auch Meerestiere Nitrilverbindungen. Dazu gehören das Bursatellin[S 11] aus Seehasen der Gattung Bursatella[99] und die aus Schwämmen isolierten Calyculine.[100] Bei den Albanitrilen aus Schwämmen der Gattung Mycale handelt es sich um lineare Verbindungen (Kettenlänge 16 bis 18), die an einem oder beiden Enden eine Nitrilfunktion und zusätzlich mehrere C≡C-Dreifachbindungen aufweisen.[101]
Vorkommen in Pilzen
[Bearbeiten | Quelltext bearbeiten]Viele Pilze bilden Cyanwasserstoff aus Glycin. Dazu gehören Vertreter der Gattungen Trichterlinge (Clitocybe), Schwindlinge (Marasmius), Stielporlinge (Polyporus) und Ritterlinge (Tricholoma).[102] Die Epurpurine sind eine Gruppe gelber Phenolfarbstoffe, die jeweils zwei Nitrilgruppen tragen und in Emericella purpurea vorkommen.[103] Diatretin II kommt im Fleischfalben Trichterling (Clitocybe diatreta)[104] und dem violetten Rötelritterling vor.[105] Im Nelken-Schwindling kommt das Cyanhydrin der Glyoxalsäure vor, das aus zwei Molekülen Glycin gebildet wird und bei Beschädigung des Pilzes Blausäure freisetzt.[106]
Vorkommen in Bakterien
[Bearbeiten | Quelltext bearbeiten]Cyanwasserstoff wird von diversen Bodenbakterien gebildet, darunter Cyanobakterien und Vertreter der Gattungen Aeromonas, Bacillus und Pseudomonas. Die Biosynthese erfolgt ausgehend von Glycin.[107] Aus Pseudomonas veronii wurde eine Gruppen von Alkannitrilen isoliert: Dodecannitril, Tridecannitril,[S 12] Tetradecannitril, Pentadecannitril[S 13] und Hexadecannitril,[S 14] außerdem Verbindungen mit ähnlicher Kettenlänge, die aber eine Doppelbindung aufweisen. Aus Micromonospora echinospora wurden ebenfalls Verbindungen mit ähnlicher Kettenlänge isoliert, die aber eine Methylverzweigung am Kettenende, eine Doppelbindung, oder beides aufweisen.[108] Aus Streptomyces regensis ist ein Cyanhydrin bekannt, das zusätzlich eine Phosphonsäuregruppe aufweist.[109] Das Aethokthonotoxin aus dem Cyanobakterium Aetokthonos hydrillicola ist ein bromiertes Indolderivat, das zusätzlich eine Nitrilgruppe trägt. Es ist ein Neurotoxin, das oft zum Tod von Weißkopfseeadlern führt, die es über die Nahrung aufnehmen.[110]
Vorkommen im Weltall
[Bearbeiten | Quelltext bearbeiten]Nitrile gehören zu den häufigsten organischen Molekülen im Weltall und mehr als zehn Verbindungen wurden eindeutig nachgewiesen.[111] Cyanwasserstoff war eine der ersten mehratomigen Spezies, die im Weltall nachgewiesen wurde und kommt dort vergleichsweise häufig und in größeren Mengen vor.[112] Zu den im Weltall nachgewiesenen Nitrilen gehören außerdem Acetonitril und Aminoacetonitril,[111] sowie Isobutyronitril,[113] Cyanoacetylen und Cyanopolyine mit zwei bis fünf konjugierten Dreifachbindungen.[114] In der Atmosphäre des Saturnmondes Titan kommen Cyanwasserstoff, Cyanoacetylen und Dicyan vor.[115]
Bedeutung für die Entstehung des Lebens
[Bearbeiten | Quelltext bearbeiten]Nitrile spielten möglicherweise eine wichtige Rolle bei der Entstehung des Lebens auf der Erde.[116][115] In mehreren Experimenten konnte gezeigt werden, dass es viele mögliche Umgebungsbedingungen gibt unter denen Cyanwasserstoff entstehen kann. Ausgangsprodukte können Gasgemische aus Methan, Kohlenstoffdioxid, Stickstoff, Ammoniak und/oder Wasserstoff sein. Denkbar sind außerdem verschiedene Formen einer Energiezufuhr wie elektrische Entladungen oder UV-Strahlung. Aus Cyanwasserstoff wiederum bilden sich unter einfachen Bedingungen weitere Moleküle.[117] So werden Cyanwasserstoff und andere Nitrile wie Cyanoacetylen und Dicyan als Vorläufer der Nukleinbasen vermutet.[115][117] Aminonitrile wiederum sind wahrscheinliche Vorläufer der Aminosäuren und Peptide, zum Beispiel Aminoacetonitril von Glycin. Hierbei wird ein Prozess analog zur Strecker-Synthese vermutet, sodass sich zunächst aus Cyanid, Acetaldehyd und Ammoniak das α-Aminopropionitril bilden könnte, das anschließend zu Alanin hydrolysiert wird.[116][118][119]
Herstellung
[Bearbeiten | Quelltext bearbeiten]Für die Herstellung von Nitrilen existiert eine große Anzahl an Methoden. Dazu gehören die Kolbe-Nitril-Synthese, die Dehydratisierung von Carbonsäureamiden und Aldoximen und die Oxidation primärer Amine.
Additions- und Substitutionsreaktionen mit Cyanidgruppen
[Bearbeiten | Quelltext bearbeiten]Ein mögliches Herstellungsverfahren für Nitrile ist die Kolbe-Nitrilsynthese. Dabei entsteht in einer Substitutionsreaktion aus einem reaktionsfähigen Alkylhalogenid und einem Alkalicyanid (Natriumcyanid oder Kaliumcyanid) das Alkannitril und ein Alkalihalogenid. Die Reaktion eignet sich besonders gut zur Umsetzung primärer sowie allylischer und benzylischer Halogenide. Sekundäre Alkylhalogenide liefern schlechtere Ausbeuten, tertiäre reagieren stattdessen nur durch Eliminierung. Neben Halogeniden können auch andere Edukte mit guten Abgangsgruppen verwendet werden. Silbercyanid ist im Gegensatz zu Alkalicyaniden zur Herstellung von Nitrilen nicht als Reagenz geeignet, da mit diesem vorzugsweise Isonitrile gebildet werden.[120] Ein Beispiel für eine Kolbe-Nitrilsynthese ist die Reaktion von Methyliodid mit Natriumcyanid zu Acetonitril und Natriumiodid:[121]

Analog kann 1,3-Dibrompropan mit Natriumcyanid zu Pentandinitril umgesetzt werden[122] oder 1-Iodoctan mit Kaliumcyanid zu Nonannitril.[123] Mit Cyanwasserstoff / Triethylaluminium oder mit Diethylaluminiumcyanid können ebenfalls Cyanierungen durchgeführt werden, beispielsweise die Ringöffnung eines Epoxids zu einem β-Cyanhydrin oder die 1,4-Addition von Cyanid an ein Enon.[124][125] Trimethylsilylcyanid ist ein weiteres Cyanierungsreagenz mit dem Epoxide zu β-Cyanhydrinen geöffnet werden können. Dabei wird das Sauerstoffatom silyliert.[126] Mit Trimethylsilylcyanid gelingt außerdem die Substitution tertiärer Alkylhalogenide, die mit der Kolbe-Nitril-Synthese nicht möglich ist.[120]
Mit geeigneten Übergangsmetallkatalysatoren kann in einer Hydrocyanierung Cyanwasserstoff an die Mehrfachbindungen von Alkenen und Alkinen addiert werden, wodurch Nitrile erhalten werden. Hierzu werden meist Nickelkatalysatoren verwendet. Eine direkte Verwendung von Cyanwasserstoff ist oft nicht nötig, stattdessen eignen sich auch Syntheseäquivalente wie Acetoncyanhydrin oder Isovaleronitril.[127] Ein wichtiger industrieller Prozess ist außerdem die Hydrocyanierung von Butadien zu Adiponitril.[7]
Dehydratisierungsreaktionen
[Bearbeiten | Quelltext bearbeiten]
Carbonsäureamide und Aldoxime können durch Dehydratisierung (Abspaltung eines Wassermoleküls) zu Nitrilen umgesetzt werden, wofür eine große Zahl an Reagenzien und Methoden bekannt ist.[128][129][130] Auch für die Herstellung von Nitrilen durch die Dehydratisierung von Nitroalkanen sind Methoden bekannt.[131]
Ein Reagenz für die Dehydratisierung von Carbonsäureamiden, das schon Mitte des 19. Jahrhunderts bekannt war, ist Phosphorpentoxid.[128] Amide können außerdem durch Umsetzung mit dreiwertigen Phosphorreagenzien wie Phosphortrichlorid oder Triphenylphosphit dehydratisiert werden;[130] sowie mit Diethylchlorphosphat,[132] Thionylchlorid[133] oder Phosgen.[134] Bei Einsatz bestimmter Palladium-Komplexe oder eines anderen geeigneten Katalysators kann Acetonitril als Dehydratisierungsreagenz wirken, um ein Amid in ein Nitril zu überführen, wobei es selbst zu Acetamid reagiert. Analog kann auch Dichloracetonitril verwendet werden.[135][136] Ähnliche Methoden verwenden Eisen(II)-chlorid-Tetrahydrat, Zinktriflat oder Uranylnitrat-Hexahydrat als Katalysator und N-Methyl-N-trimethylsilyltrifluoracetamid als Dehydratisierungsreagenz.[137][138][139] Auch mittels eines Reaktionssystems aus Triphenylphosphin, Iod und N-Methylmorpholin können Carbonsäureamide dehydriert werden.[140] Eine weitere Methode stellt die Dehydratisierung bei hohen Temperaturen (220–240 °C) in Hexamethylphosphorsäuretrisamid (HMPT) dar.[141] Die Dehydratisierung von primären Amiden mit Zinkchlorid unter Einwirkung von Mikrowellen ist umkehrbar. In wässrigem Acetonitril kann ein Amid zum Nitril umgesetzt werden. In einem Wasser-THF-System mit Zusatz von Acetamid läuft hingegen die umgekehrte Reaktion von Nitril zu Amid ab.[142]
Sowohl Carbonsäureamide als auch Aldoxime können durch Umsetzung mit Aluminiumchlorid und Natriumiodid in Acetonitril dehydratisiert werden.[129] Ebenso können beide Verbindungsklassen mit Oxalylchlorid und einer katalytischen Menge Dimethylsulfoxid dehydratisiert werden, in einer Reaktion, die ähnlich einer Swern-Oxidation abläuft.[143] Auch die Umsetzung zu Nitrilen unter Katalyse mit siebenwertigem Rhenium (Perrheniumsäure oder Trimethylsilylperrhenat[S 15]) gelingt sowohl mit Amiden als auch mit Aldoximen. Als Nebenprodukt anfallendes Wasser kann durch azeotrope Destillation entfernt werden.[144]
Aldoxime können außerdem mit Cyanurchlorid dehydratisiert werden,[145] mit dem Burgess-Reagenz,[146] sowie mit einer Kombination von Trifluormethansulfonsäureanhydrid und Triphenylphosphin, wobei letzteres zu Triphenylphosphinoxid oxidiert wird.[147] Auch eine katalytische Dehydrierung ist möglich, beispielsweise mit Eisen(III)-triflat,[148][S 16] mit Kupfer(II)-acetat,[149] mit einem gemischten Hydroxid von Zinn und Wolfram[150] oder mit einem bimetallischen Palladium-Mangan-Katalysator.[151] Schließlich ist auch die enzymatische Dehydratisierung von Aldoximen mit Aldoxim-Dehydratasen möglich. Dabei handelt es sich um Enzyme, die in Bakterien vorkommen, darunter Pseudomonas chlororaphis, und die schon verschiedentlich zur Synthese von Nitrilen verwendet wurden.[152]
Herstellung aus Aldehyden und Ketonen
[Bearbeiten | Quelltext bearbeiten]
Aldehyde können mit Hydroxylaminhydrochlorid in Oxime überführt und dann weiter zu Nitrilen dehydratisiert werden (beispielsweise mit Oxalylchlorid).[153] Auch direkte Umsetzungen von Aldehyden zu Nitrilen sind bekannt, möglich ist dies mittel Hydroxylamin-O-sulfonsäure[154] oder O-(4-Trifluormethylbenzoyl)hydroxylamin.[155][S 17] Eine solche Umsetzung ist auch mit Hydroxylamin möglich, wenn Titan(IV)-chlorid oder ein gemischtes Hydroxid von Zinn und Wolfram als Katalysator verwendet wird[150][156] oder wenn Sulfurylfluorid oder Selenoxid als zusätzliches Reagenz verwendet wird.[157][158] Tosylmethylisocyanid (auch Van-Leusen-Reagenz genannt) ermöglicht die direkte Umwandlung eines Ketons in ein Nitril, was als Van-Leusen-Reaktion bezeichnet wird. Dabei wird die gesamte Nitrilgruppe und damit auch ein zusätzliches Kohlenstoffatom eingeführt.[159][160][161]
Herstellung aus Aminen
[Bearbeiten | Quelltext bearbeiten]
Primäre Amine (R-CH2-NH2) können mittels verschiedener Methoden zu Nitrilen oxidiert werden. Es sind mehrere Verfahren bekannt, die Nitroxylradikale wie TEMPO und dessen Derivat 4-Acetamido-TEMPO als katalytisches Oxidationsmittel verwenden. Diese können durch Oxone als stöchiometrisches Oxidationsmittel regeneriert werden oder elektrochemisch durch Anlegen einer Spannung.[162][163] Eine andere Methode verwendet Kupfer(I)-chlorid oder Kupfer(II)-chlorid als Katalysator, Sauerstoff als stöchiometrisches Oxidationsmittel und zusätzlich ein Molsieb zum Abfangen des entstehenden Wassers.[164]
Ammoxidation
[Bearbeiten | Quelltext bearbeiten]Die Ammoxidation ist eine heterogen katalysierte Gasphasenreaktion, in der aliphatische Verbindungen oder methylsubstituierte aromatische Verbindungen durch Umsetzung mit Sauerstoff (Luft) und Ammoniak zu Nitrilen umgesetzt werden, wobei Wasser als Nebenprodukt anfällt. Die Prozesstemperatur beträgt über 300 °C, als Katalysatoren kommen Oxide von Vanadium, Chrom oder Molybdän zum Einsatz.[165] Acrylnitril, ein wichtiger Ausgangsstoff zur Herstellung von Polymeren (siehe Abschnitt Verwendung), wird überwiegend durch Ammoxidation von Propylen hergestellt.[8] Das wichtigste Herstellungsverfahren für Cyanwasserstoff ist der Andrussow-Prozess, das heißt die Ammoxidation von Methan mit einem Platin-Katalysator. Eine erhebliche Menge des weltweit verwendeten Cyanwasserstoffs fällt allerdings auch bei der Herstellung von Acrylnitril als Nebenprodukt an.[166]
Sonstige Herstellungsverfahren
[Bearbeiten | Quelltext bearbeiten]Durch die Carbocyanierung kann ein Nitril an eine Mehrfachbindung addiert werden, wobei ein weiteres Nitril erhalten wird. Arylnitrile können unter Katalyse mit Bis(cyclooctadien)nickel(0) und Trimethylphosphin an Alkine addiert werden, wodurch α,β-ungesättigte Nitrile erhalten werden. Mit gewissen Änderungen an den Reaktionsbedingungen wie der Verwendung eines anderen Phosphins oder Zusatz einer Lewis-Säure wie Trimethylaluminium oder Triphenylboran, können auch nichtaromatische Nitrile addiert werden, sowohl gesättigte als auch α,β-ungesättigte.[167] Es sind auch Carbocyanierungen bekannt, bei denen zwei Moleküle verbunden werden und zusätzlich eine Nitrilgruppe eingeführt wird. Dies funktioniert durch Einsatz von Hexabutyldistannan sowie von Tosylcyanid[S 18] als Quelle der Cyanidgruppe.[168]
Carbonsäuren können durch Umsetzung mit Indium(III)-chlorid in Acetonitril bei 200 °C in die entsprechenden Nitrile überführt werden. Dabei wirkt Acetonitril sowohl als Lösungsmittel als auch als Quelle für Stickstoffatome und wird bei der Reaktion zu Essigsäure umgesetzt. Die Reaktion verläuft über mehrere Mumm-Umlagerungen.[169] Alkohole können durch eine Mitsunobu-Reaktion in Nitrile überführt werden. Dazu kann Cyanomethylidentrimethylphosphoran[S 19] in Gegenwart von Acetoncyanhydrin verwendet werden.[170] N-Alkylamide können im Von-Braun-Abbau durch Umsetzung mit Phosphorpentachlorid in Nitrile umgewandelt werden.[171] Alternative Reagenzien sind Phosphorpentabromid und Carbonylbromid.[172]
Herstellung von aromatischen Nitrilen
[Bearbeiten | Quelltext bearbeiten]
Die Herstellung von Arylnitrilen gelingt unter anderem durch die Sandmeyer-Reaktion von Aryldiazoniumsalzen mit Kupfer(I)-cyanid)[173] und durch die Rosenmund-von Braun-Reaktion (direkte Umsetzung eines Arylbromids mit Kupfer(I)-cyanid).[174] Umsetzung von Metallthiocyanaten mit aromatischen Carbonsäuren wird als Letts-Nitrilsynthese bezeichnet. Dafür kann Kaliumthiocyanat verwendet werden, aber Bleithiocyanat ergibt bessere Ergebnisse.[3]
Aryliodide können unter Palladiumkatalyse mit Trimethylsilylcyanid zu aromatischen Nitrilen umgesetzt werden. So ergibt die Reaktion von Iodbenzol mit Trimethylsilylcyanid und Tetrakis(triphenylphosphin)palladium(0) (Pd(PPh3)4) als Produkt Benzonitril.[175] Eine weitere Synthesemöglichkeit, die ebenfalls auf Palladiumkatalyse beruht (auch hierfür kann Pd(PPh3)4 eingesetzt werden) ist die Decarbonylierung aromatischer Acylcyanide.[176] Auch die palladiumkatalysierte Cyanierung von Arylchloriden mit Kaliumcyanid[177] oder Kaliumhexacyanidoferrat(II)[178] ist bekannt. Chinone können mit Trimethylsilylcyanid zu silylierten Cyanhydrinen umgesetzt werden und anschließend mit Phosphortribromid aromatisiert werden.[179] Eine weitere mögliche Synthese von Arylnitrilen ist die Umsetzung von Arylgrignard-Verbindungen oder Aryllithium-Verbindungen mit Dimethylmalonitril.[180][S 20]
Herstellung von Cyanhydrinen
[Bearbeiten | Quelltext bearbeiten]
Cyanhydrine können durch Addition eines Alkalicyanids an einen Aldehyd oder ein Keton in Gegenwart von Essigsäure hergestellt werden. Für Substrate, die nach dieser Methode nicht reagieren, eignet sich Diethylaluminiumcyanid als Alternative. Eine andere Methode ist die Transhydrocyanierung, bei der Cyanwasserstoff von Acetoncyanhydrin auf ein Aldehyd oder Keton übertragen wird.[5] Als Katalysatoren für letztere Reaktion eignen sich Alkoholate von Lanthaniden wie Lanthan(III)-isopropanolat,[S 21] Cer(III)-isopropanolat,[S 22] Samarium(III)-isopropanolat[S 23] und Ytterbium(III)-isopropanolat.[181][S 24]
Bei der Addition von Trimethylsilylcyanid an Ketone oder Aldehyde entstehen Cyanhydrine als Trimethylsilylether.[182][183] Als Katalysatoren eignen sich Zinkiodid, Kaliumcyanid mit [18]Krone-6 oder Ytterbium(III)-cyanid.[5][S 25] Unter geeigneten Bedingungen können solche Reaktionen enantioselektiv erfolgen. Hierzu eignen sich Vanadium- oder Titankatalysatoren, die einen chiralen, Salen-ähnlichen Liganden tragen. Eine andere Möglichkeit ist die Verwendung von Titan(IV)-isopropanolat und einem chiralen Imin.[184][185]
Herstellung von Acylcyaniden
[Bearbeiten | Quelltext bearbeiten]Acylcyanide beziehungsweise α-Oxonitrile können in einigen Fällen durch Umsetzung von Carbonsäurehalogeniden mit Übergangsmetallcyaniden (zum Beispiel Kupfercyanid oder Silbercyanid) hergestellt werden. Dies funktioniert besonders gut mit aromatischen Carbonsäurehalogeniden und mit Carbonsäurebromiden, während aliphatische Carbonsäurechloride gar nicht reagieren. Aliphatische Acylcyanide lassen sich durch Reaktion von Carbonsäurechloriden mit Trimethylsilylcyanid herstellen.[186]
Enantioselektive Synthese chiraler Nitrile
[Bearbeiten | Quelltext bearbeiten]Unter Rückgriff auf Chiral-Pool-Verbindungen können durch enantioselektive Synthese α-chirale nitrilhaltige Wirkstoffe in eutomerer Form, wie Vildagliptin und Saxagliptin, erhalten werden. Dabei können standardmäßige Reaktionen zur Erzeugung der Nitrilfunktion angewandt werden. So kann ein enantiomerenreines Amid oder Oxim hergestellt werden, beispielsweise aus der natürlich enantiomerenrein vorkommenden Aminosäure Prolin, und dieses dehydratisiert werden. Wie zweckmäßig ein solches Verfahren ist, hängt allerdings vom jeweiligen Zielmolekül ab. Daneben sind auch asymmetrische Cyanierungsreaktionen bekannt.[187] Wichtig ist die asymmetrische Hydrocyanierung von Carbonylverbindungen, siehe hierzu den Abschnitt Herstellung von Cyanhydrinen. Daneben sind auch viele asymmetrische Hydrocyanierungen von Iminen bekannt, die zu enantiomerenreinen α-Aminonitrilen führen.[185]
Reaktionen
[Bearbeiten | Quelltext bearbeiten]Die Nitrilfunktion hat eine große Bedeutung in der organischen Synthese, da sie einerseits über ein nucleophiles Stickstoffatom und ein elektrophiles Kohlenstoffatom verfügt, andererseits aber auch Komplexe bilden kann.[188]
Hydrolyse
[Bearbeiten | Quelltext bearbeiten]Durch Hydrolyse von Nitrilen kann man Carbonsäuren[189] herstellen. Die Hydrolyse kann sowohl sauer als auch basisch erfolgen, in beiden Fällen sind aber in der Regel drastische Bedingungen erforderlich. Bei der basischen Hydrolyse addiert sich ein Hydroxidion an das Nitril und bildet eine Imidoverbindung. Bei der sauren Hydrolyse wird das Nitril zunächst protoniert und dann unter Addition von Wasser ebenfalls eine Imidoverbindung gebildet. Die Imidoverbindung lagert sich in beiden Fällen zu einem Carbonsäureamid um, das unter den jeweiligen Bedingungen zu einer Carbonsäure weiterreagieren kann.[14]
![{\displaystyle \mathrm {{R\!\!-\!\!C\!\!\equiv \!\!N}+2~H_{2}O~~~{\overset {\Delta }{\xrightarrow[{OH^{-}}]{}}}~~~R\!\!-\!\!COOH+NH_{3}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/72b662c8fcae0924a4dd1b2fc2d205d06ab05a54)

Es sind auch Enzyme bekannt, die als Nitrilasen bezeichnet werden und die direkte Hydrolyse von Nitrilen zu Carbonsäuren ohne Amide als Zwischenstufe katalysieren.[190] Nitrile treten auch als Zwischenstufe bei der Strecker-Synthese auf. Hierbei wird ein Aldehyd mit Ammoniak und Cyanid umgesetzt, wobei zunächst ein α-Aminonitril entsteht. Dessen Hydrolyse ergibt eine α-Aminosäure. Wegen der Bedeutung der Aminosäuren sowohl in der Natur als auch in der Industrie, hat die Strecker-Synthese eine enorme historische Bedeutung, ist aber auch heute noch wichtig.[191]
Nitrile können auch gezielt zu Amiden statt zu Carbonsäuren hydrolysiert werden, sowohl im Sauren als auch im Basischen. Dazu gehört die Hydrolyse mittels Trifluoressigsäure mit Schwefelsäure beziehungsweise Essigsäure mit Schwefelsäure,[192] die Hydrolyse mit Wasser und Trimethylsilylchlorid,[193] oder mit Bortrifluorid und wässriger Essigsäure.[194] Basisch (hydroxid-katalysiert) ist die wässrige Hydrolyse mit Rutheniumhydroxid auf Alumina als Katalysator möglich,[195] und die Umsetzung mit Natriumhydroxid in Isopropanol.[196] Die Enyzme, die die Hydrolyse von Nitrilen zu Amiden katalysieren, werden als Nitrilhydratasen bezeichnet.[197]
Reduktion
[Bearbeiten | Quelltext bearbeiten]Nitrile können mit verschiedenen Reagenzien reduziert werden. Durch katalytische Hydrierung oder Umsetzung mit starken Hydridüberträgern wird so ein primäres Amin erhalten:

Ein konkretes Beispiel ist hier Lithiumaluminiumhydrid.[14] Weitere Reaktionssysteme, durch die Nitrile zu Aminen reduziert werden können, sind Raney-Nickel mit Kaliumborhydrid,[198] Raney-Nickel mit Hydraziniumformiat,[199][S 26] Raney-Cobalt,[200] Dimethoxyboran[S 27] mit Dinickelborid[201] und Rhodium auf Aluminiumoxid.[202] Die Hydrierung von Adiponitril zu Hexamethylendiamin ist ein wichtiger industrieller Prozess.[203]
Mit bestimmten Reagenzien wie Diisobutylaluminiumhydrid läuft die Reaktion nur bis zum Imin, durch dessen Hydrolyse ein Aldehyd erhalten werden kann:[14]
![{\displaystyle \mathrm {{R\!\!-\!\!C\!\!\equiv \!\!N}~~{\overset {\text{DIBAL}}{\xrightarrow {} }}~~R\!\!-\!\!CH\!\!=\!\!NH~~{\overset {\text{[H]}}{\xrightarrow {} }}~~R\!\!-\!\!CH\!\!=\!\!O} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/0fc02dd7075b8cf8e55f4777395665da842f0d06)
Auch mittels der Stephen-Reduktion können aus Nitrilen Aldehyde erhalten werden. Dazu wird das Nitril mit Zinn(II)-chlorid und Chlorwasserstoff in Ether umgesetzt.[204]
Additionen von Kohlenstoff-Nucleophilen
[Bearbeiten | Quelltext bearbeiten]Durch Addition von metallorganischen Verbindungen an Nitrile, darunter mit Grignard-Reagenzien, entstehen Iminoverbindungen, deren Hydrolyse Ketone ergibt.[14] Die Addition sterisch gehinderter Verbindungen an Nitrile kann durch Zusatz von Kupfer(I)-bromid deutlich beschleunigt werden. Ein Beispiel ist die Umsetzung von Benzonitril mit tert-Butylmagnesiumchlorid,[S 28] die ohne einen solchen Zusatz praktisch keinen Umsatz ergibt, mit dem Zusatz jedoch über 90 % innerhalb weniger Stunden.[205] Bei der Blaise-Reaktion wird aus einem α-Bromester wie Ethylbromacetat ein Zinkenolat gebildet und dieses dann an ein Nitril addiert. Durch Hydrolyse wird ein β-Ketoester erhalten.[206]
Addition von O- und N-Nucleophilen
[Bearbeiten | Quelltext bearbeiten]In der Pinner-Reaktion werden unter saurer Katalyse Alkohole an Nitrile addiert, wodurch nach Neutralisation Imidate erhalten werden.[207][208] Bei einer neueren Variante der Reaktion wird als Lösungsmittel Cyclopentylmethylether verwendet.[209] Eine wichtige Reaktion, bei der ein Sauerstoff-Nucleophil an ein Nitril addiert wird, ist das Trichloracetimidat-Verfahren. Hierbei wird ein Saccharid (Zuckermolekül) durch Addition an Trichloracetonitril in ein Trichloracetimidat überführt und so aktiviert, was die Synthese von Oligosacchariden ermöglicht.[210][211] Eine weitere wichtige Anwendung von Imidaten ist die Overman-Umlagerung, eine Variante der Claisen-Umlagerung. Dabei wird ein Allylalkohol an Trichloracetonitril oder Trifluoracetonitril addiert. Das gebildete Imidat wird zu einem N-Allylamid umgelagert.[212] Bei der Payne-Oxidation wird durch Addition eines Hydroperoxid-Ions (aus Wasserstoffperoxid) an ein Nitril, meist Acetonitril oder Benzonitril, eine Imidopersäure gebildet. Diese eignen sich für die Epoxidierung von Alkenen, analog zu anderen Persäuren, wie meta-Chlorperbenzoesäure.[213][214] Die Addition von Aminen an Nitrile ergibt Amidine. Eine solche Reaktion gelingt unter Katalyse mit Samarium(II)-iodid[215] oder mit Yttrium(III)-triflat,[S 29] Lanthan(III)-triflat,[S 30] sowie Triflaten anderer Lanthanide.[216]
Deprotonierung und α-Funktionalisierung
[Bearbeiten | Quelltext bearbeiten]Die elektronenziehenden Eigenschaften der Nitrilgruppe ermöglichen die Herstellung von Carbanionen-Nucleophilen durch α-Deprotonierung.[28] Diese können für Alkylierungs- und Acylierungsreaktionen verwendet werden, allerdings kann die Deprotonierung auch zur Racemisierung chiraler Verbindungen führen.[217] Die Selbstkondensation von Nitrilen in Gegenwart einer Base (zum Beispiel Lithiumbis(trimethylsilyl)amid) wird als Thorpe-Reaktion bezeichnet. Sie führt zu Cyanoenaminen (üblicherweise stereoselektiv zum (E)-Isomer) und eignet sich unter anderem auch zur Herstellung von cyclischen Molekülen.[218]
Reaktionen mit Aromaten
[Bearbeiten | Quelltext bearbeiten]Die Nitrilgruppe von Arylnitrilen kann einen dirigierenden Effekt ausüben und die selektive Metallierung eines Aromaten in ortho-Position zum Nitril ermöglichen. Solche Arylmetallspezies können dann in Kupplungsreaktionen eingesetzt werden.[188]
Bei der Houben-Hoesch-Reaktion wird ein Aromat unter Säurekatalyse mit einem Nitril acyliert, ähnlich einer Friedel-Crafts-Acylierung. Nach hydrolytischer Aufarbeitung wird dabei ein Keton erhalten. Die Reaktion funktioniert insbesondere mit elektronenreichen Aromaten wie Phenol. Sie eignet sich auch für Cyclisierungen, wenn es sich um eine intramolekulare Reaktion handelt.[219]
Cyclisierungen
[Bearbeiten | Quelltext bearbeiten]Nitrile können zu Triazinen trimerisiert werden. Bei Propionitril, Valeronitril oder Benzonitril, gelingt die Trimerisierung durch hohen Druck, bei 100–125 °C und etwa 7 bis 8,5 kbar in Methanol.[220] Derartige Trimerisierungen gelingen außerdem mittels Katalyse durch Samarium(II)-iodid,[221] Yttrium(III)-triflat oder einer Lewissäure (Zinkchlorid, Aluminiumchlorid oder Titanchlorid) auf Silica.[222] Durch Umsetzung von Cyanwasserstoff mit Chlor und anschließende Trimerisierung wird Cyanurchlorid hergestellt.[223] Mit Trifluormethansulfonsäureanhydrid können Triazine aus zwei verschiedenen Nitrileinheiten gebildet werden, von denen eine zweimal und eine einmal in den Ring eingebaut wird.[224] Die Polymerisation von Terephthalonitril (1,4-Dicyanobenzol) in flüssigem Zinkchlorid bei 400 °C ergibt ein zweidimensionales Netz aus Phenylen- und Triazinringen.[225]
[4+2]-Cycloaddition von Nitrilen mit Dienen sind kaum bekannt und von untergeordneter Bedeutung. Die [2+2+2]-Cycloaddition eines Nitrils mit zwei Alkinen wird durch Nickelverbindungen katalysiert und ermöglicht die Herstellung stark substituierter Pyridine.[188] Auch Reaktionen mit Cobalt und Zirconium als Katalysatoren sind bekannt.[226][227] Die [3+2]-Cycloaddition von Nitrilen mit Aziden beziehungsweise Stickstoffwasserstoffsäure ergibt Tetrazole.[188][228] Die Gewald-Reaktion ist eine Methode für die Herstellung von 2-Aminothiophenen. Hierzu wird ein Nitril mit einer zusätzlichen elektronenziehenden Gruppe (wie im Malonitril oder Methylcyanoacetat) mit elementarem Schwefel und einem Aldehyd oder Keton umgesetzt. Diese Reaktion wird für die Herstellung des Wirkstoffs Olanzepin verwendet.[229]
Weitere Reaktionen
[Bearbeiten | Quelltext bearbeiten]Die reduktive Decyanierung ermöglicht die Entfernung einer Nitrilgruppe, wenn diese lediglich wegen ihrer dirigierenden oder elektronenziehenden Eigenschaften verwendet wurde. Die klassische Methode zur reduktiven Decyanierung ist die Verwendung von elementaren Alkalimetallen mit einem Protonendonator. Mögliche Kombinationen sind Natrium in Ammoniak oder Lithium in Ethylamin.[28] Ein weiteres Beispiel ist die Verwendung von Kalium auf Aluminiumoxid in Hexan.[230] Aromatische Nitrile können auch mit Übergangsmetallkatalysatoren auf der Basis von Nickel oder Cobalt defunktionialisiert werden.[28]
Bei der Ritter-Reaktion wird ein Alken oder ein tertiärer Alkohol mit einem Nitril und Schwefelsäure umgesetzt, wodurch ein N-Alkylamid gebildet wird, wobei das Alken oder der Alkohol den Substituenten am Stickstoff ergibt.[231]
Die Protonierung oder Alkylierung von Nitrilen ergibt Nitrilium-Ionen. Die Protonierung von Nitrilen ist ein Teilschritt der sauren Hydrolyse von Nitrilen.[14] Die N-Alkylierung von Nitrilen ergibt Alkylnitrilium-Verbindungen. Dies ist möglich durch Umsetzung mit Chlorameisensäureestern in Gegenwart von Antimon(V)-chlorid.[232] N-Ethylnitriliumverbindungen entstehen durch Umsetzung von Nitrilen mit Triethyloxoniumtetrafluoroborat.[233]
Nitrile als Komplexliganden
[Bearbeiten | Quelltext bearbeiten]
Nitrile bilden diverse Komplexe mit Übergangsmetallen. Sie sind dabei im Allgemeinen über das nichtbindende Elektronenpaar des Stickstoffatoms koordiniert, es sind aber auch einige Komplexe bekannt, bei denen die Koordination über die Dreifachbindung erfolgt. Viele als Feststoff isolierbare Komplexe sind von Kupfer bekannt.[234] Andere Metalle, die Nitrilkomplexe bilden, sind unter anderem Chrom, Mangan, Zink, Niob, Rhenium und Eisen. Typische Nitrilliganden sind Acetonitril oder Benzonitril.[234][235] Hergestellt werden können Nitrilkomplexe unter anderem durch Dehydratisierung eines Hydratsalzes mit dem gewünschten Gegenion des Komplexes. Dies funktioniert mit Tetrafluoroboraten von Mangan, Eisen, Cobalt, Nickel und Kupfer. Eine andere Möglichkeit ist die Oxidation elementarer Metalle mit einem Nitrosoniumsalz mit dem gewünschten Gegenion. Ein Beispiel hierfür ist die Umsetzung von Kupfer mit Nitrosoniumperchlorat[S 31] in Acetonitril zu Tetrakis(acetonitril)kupfer(II)-perchlorat, wobei Stickstoffmonoxid als Nebenprodukt entsteht.[235]
Nitrilliganden binden in der Regel nur schwach an das Zentralatom des Komplexes und können leicht ausgetauscht werden, um andere Komplexe herzustellen.[235] Ein Beispiel ist die Umsetzung von Tetrakis(acetonitril)kupfer(I)-tetrafluoroborat zu Tetrakis(methoxyisobutylisonitril)kupfer(I)-tetrafluoroborat, das im medizinischen Bereich verwendet wird.[236] Viele Nitril-Komplexe haben katalytische Eigenschaften in der Polymerisation von Cyclopentadien und anderen Reaktionen. Tetrakis(acetonitril)kupfer(I)-perchlorat katalysiert Michaeladditionen an Enone wie die Alkylierung von Cyclohexenon mit Diethylzink.[235]
Verwendung
[Bearbeiten | Quelltext bearbeiten]Cyanwasserstoff wird in der chemischen Industrie in großer Menge als Intermediat zur Herstellung anderer Verbindungen eingesetzt. Acrylnitril ist ein wichtiger Ausgangsstoff zur Herstellung von Nitril-Polymeren. Acetonitril ist ein wichtiges Lösungsmittel. Weitere Nitrile werden als Duftstoffe, Pestizide und chemische Reagenzien eingesetzt. Außerdem spielt die Nitrilgruppe eine wichtige Rolle in der Entwicklung von pharmazeutischen Wirkstoffen.
Verwendung von Cyanwasserstoff
[Bearbeiten | Quelltext bearbeiten]
Cyanwasserstoff ist eine Massenchemikalie, die weltweite Produktion betrug 2001 etwa 2,6 Millionen Tonnen. Wichtige Folgeprodukte, die daraus hergestellt werden, sind insbesondere Adiponitril, Acetoncyanhydrin, Natriumcyanid und Cyanurchlorid.[166][223] Auch Chelatbildner werden ausgehend von Cyanwasserstoff produziert,[223] beispielsweise EDTA aus Formaldehyd, Ethylendiamin, Cyanwasserstoff und Natriumhydroxid.[237] Ein wichtiger industrieller Produktionsprozess für Aminosäuren ist die Strecker-Synthese, bei der Cyanwasserstoff als Edukt verwendet wird.[238] Eine mengenmäßig wichtige Aminosäure ist Methionin, das aus Acrolein, Cyanwasserstoff und Schwefelwasserstoff hergestellt und insbesondere in Tierfutter verwendet wird.[223][239]
Kunststoffherstellung
[Bearbeiten | Quelltext bearbeiten]Einige weitverbreitete Polymere enthalten Acrylnitril als Monomer und weisen daher Nitrilgruppen auf. Reines Polyacrylnitril (PAN) lässt sich schlecht verarbeiten, weshalb bei der Herstellung von Polyacrylnitril neben 85 bis 99 % Acrylnitril fast immer kleine Mengen anderer Monomere zugesetzt werden.[8] Verwendet werden daneben auch Copolymere, die zwischen 35 und 85 % Acrylnitril als Monomer enthalten, neben anderen Monomeren wie Vinylacetat und Methylmethacrylat.[8][240] Die Nitril-Polymere sind neben Polyestern und Polyamiden eines der wichtigsten vollsynthetischen Materialien für Textilfasern.[8][241] Diese Fasern werden als Acrylfasern bezeichnet und in einer Größenordnung von mehreren Millionen Tonnen pro Jahr hergestellt. Im Jahr 2000 betrug die weltweite Produktion etwa 2,7 Millionen Tonnen.[8][242] Verwendet werden Acrylfasern unter anderem in Bekleidung (Socken und Pullover) sowie für Decken, Teppiche und Strickgarn.[8][243] PAN ist außerdem der wichtigste Ausgangsstoff zur Herstellung von Carbonfasern, die als extrem leichtes aber gleichzeitig stabiles Material beim Bau von Autos und Flugzeugen verwendet werden.[8][240][244][245] Die weltweite Produktionsmenge des Monomers Acrylnitril betrug 1988 etwa 3,2 Millionen Tonnen.[166]
Acrylnitril-Butadien-Copolymere werden als Nitrilkautschuk bezeichnet und haben verschiedene vorteilhafte Eigenschaften wie hohe Zugfestigkeit und Abriebfestigkeit, sowie Beständigkeit gegen Kohlenwasserstoffe (Öl und Treibstoffe). Sie werden daher für Dichtungsringe und Öl- und Treibstoffschläuche verwendet.[8] Eine weitere wichtige Anwendung von Nitrilkautschuk sind Schutzhandschuhe, die oft statt Handschuhen aus Latex im Gesundheitswesen verwendet werden, da letztere regelmäßig Allergien verursachen.[246] Solche Handschuhe werden außerdem oft bei der Arbeit mit gefährlichen Chemikalien wie organischen Lösungsmitteln verwendet.[247]
Ein weiteres wichtiges Polymer ist das Terpolymer aus Acrylnitril, Butadien und Styrol (ABS). Dieses ist eines der wichtigsten Materialien für Außenhüllen von elektronischen Geräten (Computer, Bildschirme und Tastaturen).[248] Weitere Verwendungen sind Plastikteile von Autos (an Scheinwerfern und Spiegeln); Einsätze für Kühlschränke; Außenhüllen von Küchengeräten, Staubsaugern und Elektrowerkzeugen; sowie Koffer, Vesperdosen.[249] und Spielzeuge, darunter Lego-Steine.[250][251] Auch ABS wird in der Größenordnung von mehreren Millionen Tonnen pro Jahr produziert, beispielsweise etwa 2,7 Millionen Tonnen im Jahr 1992.[249]
Polyamid 6.6 (Nylon) ist kein Nitril, allerdings ist ein Schlüsselintermediat zu dessen Herstellung das Adiponitril. Adiponitril wird durch Hydrocyanierung von Butadien oder durch Dimerisierung von Acrylnitril hergestellt und durch katalytische Hydrierung zu Hexamethylendiamin umgesetzt, welches eines der Monomere für Nylon ist. Das zweite Monomer, Adipinsäure wird durch Oxidation von Cyclohexan gewonnen.[252][253] Acetoncyanhydrin ist ein wichtiges Intermediat zur Herstellung von Methylmethacrylat, welches wiederum zur Herstellung von Polymethylmethacrylat verwendet wird.[254]
-
 Polyacrylnitril wird in Strickgarn verwendet
Polyacrylnitril wird in Strickgarn verwendet -
 Carbonfasern werden oft aus Polyacrylnitril hergestellt
Carbonfasern werden oft aus Polyacrylnitril hergestellt -
 Schutzhandschuhe aus Nitrilkautschuk
Schutzhandschuhe aus Nitrilkautschuk -
 Lego-Bausteine werden aus Acrylnitril-Butadien-Styrol-Copolymer (ABS) hergestellt
Lego-Bausteine werden aus Acrylnitril-Butadien-Styrol-Copolymer (ABS) hergestellt




Chemisch-pharmazeutische Industrie und Labore
[Bearbeiten | Quelltext bearbeiten]Acetonitril findet als Lösungsmittel Verwendung, insbesondere in der pharmazeutischen Industrie.[255] Im Jahr 2022 wurden laut einer Marktanalyse weltweit etwa 180.000 Tonnen Acetonitril produziert, wovon etwa 70 % in der pharmazeutischen Industrie verwendet wurden.[256] Es ist außerdem eines der wichtigsten Lösungsmittel für Analysen mittels HPLC.[255][257] Die Zersetzung von Azobisisobutyronitril (AIBN) und verwandten Verbindungen (zum Beispiel Azobiscyclohexancarbonitril[S 32]) ergibt vergleichsweise stabile Radikale, weshalb diese Verbindungen als Radikalstarter für radikalische Reaktionen, insbesondere Polymerisationen, verwendet werden.[258] Das Chinon DDQ, das über zwei Nitrilgruppen verfügt, ist ein weitverbreitetes Oxidationsmittel, das auch in der pharmazeutischen Industrie verwendet wird.[259] Nitrilgruppen lassen sich als Sonde für infrarot-spektroskopische Untersuchungen in Biomoleküle einführen.[260] Einige Nitrile werden als Edukte für die Synthese von Pharmazeutika verwendet.[27] Ketoprofen ist ein in einigen EU-Ländern zugelassenes Antiphlogistikum, für dessen industrielle Synthese Propionitril verwendet wird.[261][262]
Nitrile in der Medizin
[Bearbeiten | Quelltext bearbeiten]Nitrile finden sich in einem breiten Spektrum an Arzneistoffgruppen. Zwischen 2010 und 2020 wurde jedes Jahr mindestens ein Arzneistoff mit Nitrilfunktion von der amerikanischen Food and Drug Administration zugelassen. Die Nitrilgruppe hat charakteristische physikalische und chemische Eigenschaften, die für die Gestaltung von Wirkstoffen eine wichtige Rolle spielen. Als strukturelles Element weist diese Gruppe eine lineare Geometrie auf und beansprucht nur sehr wenig Raum, etwa ein Achtel im Vergleich zu einer Methylgruppe. Damit ist sie als Strukturbestandteil von Liganden gut geeignet, Hohlräume einer Bindungstasche eines Zielproteins aufzufüllen, die entsprechend schlank und tief sind und anderweitig kaum zu besetzen sind. Der Einbau einer Nitrilgruppe in ein Molekül führt im Allgemeinen zur Senkung seines Octanol-Wasser-Verteilungskoeffizienten bzw. zur Erhöhung seiner Wasserlöslichkeit. Dies wirkt sich bei an sich lipophileren Verbindungen in der Regel günstig auf die Bioverfügbarkeit, die Plasmahalbwertszeit und damit auf deren Wirkungsdauer aus. Die Nitrilgruppe in Arzneistoffen ist zumeist metabolisch sehr stabil.[263] Die Nitrilgruppe ist isoster zur Carbonylgruppe, der Hydroxylgruppe und zum Chloratom. Sie hat demnach ähnliche elektronische und sterische Eigenschaften wie diese und kann gegen diese ausgetauscht werden, um die Moleküleigenschaften leicht zu variieren.[264]
Die Wasserstoffbrückenbindung ist das wichtigste pharmakodynamische Bindungsmuster der Nitrilgruppe, die aufgrund der Elektronegativität ihres Stickstoffatoms, im Gegensatz zur Ethinylgruppe, als Protonenakzeptor fungiert.[265] So bildet beispielsweise die Nitrilgruppe des kompetitiven PDE3-Inhibitors Milrinon über einen an der Ligandenbindungsstelle dieser Phosphodiesterase befindlichen Histidin-Rest eine affinitätsrelevante Wasserstoffbrücke.[263] Nitrile sind fähig, eine koordinative Bindung mit Calciumkationen einzugehen, was für die Wirkung von Calciumkanalblockern des Verapamil-Typs essentiell ist. Diese hemmen den Calciumstrom, indem der Ligand-Calcium-Komplex eine Salzbrücke eingeht mit einem der Glutamatreste des Selektivitätsfilters in der Pore dieses ionotropen Rezeptors.[266][267] Eingesetzt wird Verapamil bei Herz-Kreislauf-Erkrankungen wie arterieller Hypertonie und Angina pectoris.[268]
Nitrilsubstituenten verringern durch starken Elektronenzug die Elektronendichte von Aromaten. Moduliert werden auf diese Weise π-π-Wechselwirkungen zwischen Wirkstoff und geeigneten Aminosäureresten eines Zielproteins, wie Phenylalanin, Tyrosin, Tryptophan und Histidin.[264] Eine solche π-π-Wechselwirkung wird beobachtet bei den Aromatasehemmern Letrozol und Anastrozol, die als Antiestrogene wirken und bei Brustkrebs eingesetzt werden.[269][270] Viele Androgenrezeptor-Antagonisten enthalten einen ausgeprägt elektronenarmen aromatischen Ring, der für die supramolekulare Rezeptorbindung von besonderer Bedeutung ist.[271] In Bicalutamid, Enzalutamid und weiteren Analoga, die zur Behandlung von Prostatakrebs eingesetzt werden, trägt eine Nitrilgruppe zu dieser Ladungsverschiebung bei.[272]
Einige Fälle sind bekannt, in denen Nitrile eine reversible, gleichwohl wirkungsrelevante kovalente Bindung zu einem Zielmolekül ausbilden.[264] Dabei addieren unter geeigneten Bedingungen Hydroxyl- oder Thiolgruppen von Serin- oder Cysteinresten der Zielproteine mit der Nitrilgruppe zu Oxo- oder Thioimidaten. Dies gilt für den bei Diabetes mellitus Typ II eingesetzten Dipeptidylpeptidase-4-Inhibitor Vildagliptin,[273] genauso wie für Saxagliptin.[274] Das antibakterielle Antibiotikum Cefmetazol[S 33] wirkt ebenfalls als kovalenter Inhibitor, in diesem Fall einer bakteriellen Protease.[275] Für den Calcium-Sensitizer Levosimendan wird eine Reaktion mit dem Proteinkomplex Troponin-C angenommen.[276] Eine solch reaktive Gruppe wird auch als Warhead (wörtlich übersetzt „Gefechtskopf“) bezeichnet.
In einigen Fällen sind Nitrilgruppen hauptsächlich wegen ihrer Sterik (das heißt räumlichen Gestalt) relevant, indem sie Van-der-Waals-Kontakte mit Aminosäureresten ausbilden. Die trifft zu auf den Tyrosinkinase-Inhibitor Bosutinib, der bei Chronischer myeloischer Leukämie eingesetzt wird. Kristallstrukturen sind bekannt, in denen Bosutinib mit diversen Tyrosinkinasen komplexiert ist. Inhibitoren der Reversen Transkriptase, wie Etravirin und Rilpivirin, werden in Kombinationspräparaten gegen HIV eingesetzt. Die Acrylnitril-Teilstruktur des Rilpivirins taucht in einen aus Tyrosin, Phenylalanin und Tryptophan bestehenden aromatischen Käfig ein, wie die zugehörige Raumstruktur, die im Jahr 2008 veröffentlicht wurde, erkennen lässt.[277] Der Serotonin-Wiederaufnahmehemmer Citalopram, der gegen Depressionen eingesetzt wird, war im Jahr 2016 mit 290 Millionen definierten Tagesdosen das meist verschriebene Psychopharmakon in Deutschland. Die Nitrilgruppe des Escitaloprams zeichnet sich durch optimale Komplementarität gegenüber der zentralen und einer weiteren allosterischen Bindungsstelle des Transportmoleküls aus, wie es aus der Kristallstruktur hervorgeht.[278]
-
 Levosimendan
Levosimendan -
 Letrozol
Letrozol -
 Saxagliptin
Saxagliptin -
 Milrinon
Milrinon -
 Cefmetazol
Cefmetazol -
 Citalopram
Citalopram -
 Rilpivirin
Rilpivirin







Sonstige Verwendungen
[Bearbeiten | Quelltext bearbeiten]
Eine zweistellige Anzahl an Nitrilen wird als Duftstoffe für Kosmetika verwendet. Dazu gehören unter anderem Zimtsäurenitril, Dodecannitril, Benzonitril und Geranylnitril.[279][280][281] Nitrile weisen zum Teil ähnliche Düfte auf wie entsprechende Aldehyde, sind aber deutlich stabiler, sodass sie sich als Ersatz eignen. So eignet sich Geranylnitril als Zitrusduft und ist im Gegensatz zum strukturanalogen Citral stabil gegen Oxidation.[282]
Diverse Nitrile finden als Pestizide Verwendung. Cyanogruppen sind in einigen Pyrethroid-Insektiziden enthalten. Pyrethroide sind Carbonsäureester und durch Verwendung von 3-Phenoxymandelonitril als Alkoholkomponenten, wie in Deltamethrin und Cypermethrin, wurde eine Gruppe besonders wirksamer Derivate entwickelt.[283] Azoxystrobin war schon 1999, wenige Jahre nach Einführung, mit über 400 Millionen Dollar Verkaufswert das meistverkaufte Agrar-Fungizid und hat seine Bedeutung auch mehr als 15 Jahre später noch behalten, sodass es auch 2016 noch das meistverkaufte Fungizid war.[284][285] Azoxystrobin wurde ausgehend von dem natürlich vorkommenden Strobilurin A entwickelt. Wichtige Unterschiede zwischen der Startverbindung und dem fertigen Wirkstoff sind der Austausch der Doppelbindungen gegen aromatische Ringe und die Einführung einer Cyanogruppe an dem Ring, der schon in der Ausgangsverbindung enthalten ist.[285] Ein viel genutztes Nitril-Insektizid ist Fipronil.[286]


Cyanacrylate werden als Sekundenkleber verwendet, da sie als Einkomponentenzubereitung schnell bei normalen Umgebungsbedingungen aushärten und viele Materialien kleben können. Die bei weitem meistgenutzte Verbindung in diesem Bereich ist Ethylcyanacrylat, in geringerem Maße werden Methylcyanacrylat und Allylcyanacrylat verwendet.[287] Cyanacrylatkleber werden auch im medizinischen Bereich verwendet, um Wunden zu verkleben, anstatt sie zu nähen. Da Ester mit kurzkettigen Alkylresten (wie Methylcyanacrylat) aber oft Nebenwirkungen, insbesondere Entzündungen, verursachen, werden hier andere Verbindungen verwendet als im technischen Bereich. So kommen vor allem Butylcyanacrylat und 2-Octylcyanacrylat[S 34] zum Einsatz.[9]
Nitrile finden Verwendung als Elektrolytzusatzmittel in Lithiumbatterien. So bewirkt der Zusatz von 1,3,6-Hexantricarbonitril[S 35] eine signifikante Zunahme der Leistung gegenüber einer entsprechenden Batterie ohne Zusatz. Die Wirkweise der Nitrilzusätze ist noch nicht vollständig geklärt.[288][289]
Literatur
[Bearbeiten | Quelltext bearbeiten]- David T. Mowry: The Preparation of Nitriles. In: Chemical Reviews. Band 42, Nr. 2, 1. April 1948, S. 189–283, doi:10.1021/cr60132a001.
- Camille Scotti, James W. Barlow: Natural Products Containing the Nitrile Functional Group and Their Biological Activities. In: Natural Product Communications. Band 17, Nr. 5, Mai 2022, doi:10.1177/1934578X221099973.
- Mohammed H. Al‐Huniti, Mitchell P. Croatt: Metal-Catalyzed Dehydration of Primary Amides to Nitriles. In: Asian Journal of Organic Chemistry. Band 8, Nr. 10, Oktober 2019, S. 1791–1799, doi:10.1002/ajoc.201900343.
- Bruce N. Storhoff, Huntley C. Lewis Jr.: Organonitrile complexes of transition metals. In: Coordination Chemistry Reviews. Band 23, Nr. 1, Juni 1977, S. 1–29, doi:10.1016/S0010-8545(00)80329-X.
- Xi Wang, Yuanxun Wang, Xuemin Li, Zhenyang Yu, Chun Song, Yunfei Du: Nitrile-containing pharmaceuticals: target, mechanism of action, and their SAR studies. In: RSC Medicinal Chemistry. Band 12, Nr. 10, 2021, S. 1650–1671, doi:10.1039/D1MD00131K, PMID 34778767, PMC 8528211 (freier Volltext).
Weblinks
[Bearbeiten | Quelltext bearbeiten]Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ T. K. Brotherton, J. W. Lynn: The Synthesis And Chemistry Of Cyanogen. In: Chemical Reviews. Band 59, Nr. 5, 1. Oktober 1959, S. 841–883, doi:10.1021/cr50029a003.
- ↑ F. Wöhler, J. Liebig: Untersuchungen über das Radikal der Benzoesäure. In: Annalen der Pharmacie. Band 3, Nr. 3, Januar 1832, S. 249–282, doi:10.1002/jlac.18320030302.
- ↑ a b c David T. Mowry: The Preparation of Nitriles. In: Chemical Reviews. Band 42, Nr. 2, 1. April 1948, S. 189–283, doi:10.1021/cr60132a001.
- ↑ Arthur Lapworth: XCVI.—Reactions involving the addition of hydrogen cyanide to carbon compounds. In: J. Chem. Soc., Trans. Band 83, Nr. 0, 1903, S. 995–1005, doi:10.1039/CT9038300995.
- ↑ a b c d e Robert J. H. Gregory: Cyanohydrins in Nature and the Laboratory: Biology, Preparations, and Synthetic Applications. In: Chemical Reviews. Band 99, Nr. 12, 8. Dezember 1999, S. 3649–3682, doi:10.1021/cr9902906.
- ↑ Encyclopedia of chemical processing and design. 2: Additives to alpha. Dekker, New York 1977, ISBN 978-0-8247-2452-8.
- ↑ a b Ji Yang, Peng Wang, Helfried Neumann, Ralf Jackstell, Matthias Beller: Industrially applied and relevant transformations of 1,3-butadiene using homogeneous catalysts. In: Industrial Chemistry & Materials. Band 1, Nr. 2, 2023, S. 155–174, doi:10.1039/D3IM00009E.
- ↑ a b c d e f g h i Polyacrylonitrile. In: Handbook of Thermoplastics. CRC Press, 2016, ISBN 978-0-429-10162-5, S. 157–186.
- ↑ a b David García Cerdá, Antonio Martín Ballester, Alicia Aliena-Valero, Anna Carabén-Redaño, José M. Lloris: Use of cyanoacrylate adhesives in general surgery. In: Surgery Today. Band 45, Nr. 8, August 2015, S. 939–956, doi:10.1007/s00595-014-1056-4.
- ↑ Hans Beyer und Wolfgang Walter: Organische Chemie, S. Hirzel Verlag, Stuttgart, 22. Auflage, 1991, ISBN 3-7776-0485-2, S. 266–269.
- ↑ Eintrag zu nitriles. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.N04151 – Version: 2.3.3.
- ↑ Eintrag zu carbonitriles. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.C00838 – Version: 2.3.3.
- ↑ a b Karl-Heinz Hellwich: Chemische Nomenklatur: die systematische Benennung organisch-chemischer Verbindungen ; ein Lehrbuch für Pharmazie- und Chemiestudenten. 3., überarb. Auflage. Govi-Verl, Eschborn 1998, ISBN 978-3-7741-1095-3.
- ↑ a b c d e f g h Kurt Peter C. Vollhardt, Neil Eric Schore: Organische Chemie. Hauptbd. 5. Auflage. Wiley-VCH, Weinheim 2011, ISBN 978-3-527-32754-6.
- ↑ G. P. Moss, P. A. S. Smith, D. Tavernier: Glossary of class names of organic compounds and reactivity intermediates based on structure (IUPAC Recommendations 1995). In: Pure and Applied Chemistry. Band 67, Nr. 8-9, 1. Januar 1995, S. 1307–1375, doi:10.1351/pac199567081307.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.c01485.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.t06353.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.s05573.
- ↑ M. Prabhath, Luke Williams, Shreesha Bhat, Pallavi Sharma: Recent Advances in Cyanamide Chemistry: Synthesis and Applications. In: Molecules. Band 22, Nr. 4, 12. April 2017, S. 615, doi:10.3390/molecules22040615, PMID 28417938, PMC 6154562 (freier Volltext).
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.n04150.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.n04152.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.n04148.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.n04156.
- ↑ The IUPAC Compendium of Chemical Terminology: The Gold Book. 4. Auflage. International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC 2019, doi:10.1351/goldbook.n04153.
- ↑ Elisheva Goldstein, Buyong Ma, Jenn-Huei Lii, Norman L. Allinger: Molecular mechanics calculations (MM3) on nitriles and alkynes. In: Journal of Physical Organic Chemistry. Band 9, Nr. 4, April 1996, S. 191–202, doi:10.1002/(SICI)1099-1395(199604)9:4<191::AID-POC765>3.0.CO;2-9.
- ↑ Andrew Streitwieser, Clayton H. Heathcock, Edward M. Kosower, Andrew Streitwieser: Organische Chemie. Hauptbd. 2. Auflage. Verlag Chemie, Weinheim New York 1994, ISBN 978-3-527-29005-5.
- ↑ a b c d e f Fritz Ullmann, Barbara Elvers, Stephen Hawkins, Gail Schulz: Ullmann's encyclopedia of industrial chemistry. 5th ed Auflage. VCH, Weinheim New York 1991, ISBN 978-3-527-20117-4.
- ↑ a b c d Jean-Marc R Mattalia: The reductive decyanation reaction: an overview and recent developments. In: Beilstein Journal of Organic Chemistry. Band 13, 13. Februar 2017, S. 267–284, doi:10.3762/bjoc.13.30, PMID 28326136, PMC 5331330 (freier Volltext).
- ↑ Ronaldo Pilli, Paulo Costa, Sergio Pinheiro, Peter Bakuzis: The chemistry of carbonyl compounds and derivatives. Royal Society of Chemistry, London 2022, ISBN 978-1-78801-783-1, S. 47 f.
- ↑ K. Bowden, R. Stewart: Strongly basic systems—V. In: Tetrahedron. Band 21, Nr. 3, Januar 1965, S. 261–266, doi:10.1016/S0040-4020(01)98266-3.
- ↑ Theresa Soltner, Jonas Häusler, Andreas J. Kornath: The Existence of Tricyanomethane. In: Angewandte Chemie International Edition. Band 54, Nr. 46, 9. November 2015, S. 13775–13776, doi:10.1002/anie.201506753.
- ↑ Yeon-Ran Shin, Sun-Min Jung, In-Yup Jeon, Jong-Beom Baek: The oxidation mechanism of highly ordered pyrolytic graphite in a nitric acid/sulfuric acid mixture. In: Carbon. Band 52, Februar 2013, S. 493–498, doi:10.1016/j.carbon.2012.10.001.
- ↑ O. W. Webster: Polycyanation. The Reaction of Cyanogen Chloride, Cyclopentadiene, and Sodium Hydride. In: Journal of the American Chemical Society. Band 88, Nr. 13, Juli 1966, S. 3046–3050, doi:10.1021/ja00965a028.
- ↑ Robert E. Kitson, Norman E. Griffith: Infrared Absorption Band Due to Nitrile Stretching Vibration. In: Analytical Chemistry. Band 24, Nr. 2, 18. Februar 1952, S. 334–337, doi:10.1021/ac60062a019.
- ↑ Eintrag zu Cyanwasserstoff in der GESTIS-Stoffdatenbank des IFA, abgerufen am 7. Januar 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Acetonitril in der GESTIS-Stoffdatenbank des IFA, abgerufen am 9. Dezember 2023. (JavaScript erforderlich)
- ↑ Eintrag zu Propionitril in der GESTIS-Stoffdatenbank des IFA, abgerufen am 9. Dezember 2023. (JavaScript erforderlich)
- ↑ Eintrag zu Butyronitril in der GESTIS-Stoffdatenbank des IFA, abgerufen am 9. Dezember 2023. (JavaScript erforderlich)
- ↑ Eintrag zu Isobutyronitril in der GESTIS-Stoffdatenbank des IFA, abgerufen am 9. Dezember 2023. (JavaScript erforderlich)
- ↑ Eintrag zu Acrylnitril in der GESTIS-Stoffdatenbank des IFA, abgerufen am 9. Dezember 2023. (JavaScript erforderlich)
- ↑ Eintrag zu Benzonitril in der GESTIS-Stoffdatenbank des IFA, abgerufen am 9. Dezember 2023. (JavaScript erforderlich)
- ↑ Eintrag zu Oxalsäuredinitril in der GESTIS-Stoffdatenbank des IFA, abgerufen am 2. Januar 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Malonsäuredinitril in der GESTIS-Stoffdatenbank des IFA, abgerufen am 2. Januar 2024. (JavaScript erforderlich)
- ↑ Eintrag zu Acetoncyanhydrin in der GESTIS-Stoffdatenbank des IFA, abgerufen am 20. Januar 2024. (JavaScript erforderlich)
- ↑ a b Lewis Nelson: Acute Cyanide Toxicity: Mechanisms and Manifestations. In: Journal of Emergency Nursing. Band 32, Nr. 4, August 2006, S. S8–S11, doi:10.1016/j.jen.2006.05.012.
- ↑ a b Ahmed E. Ahmed, Mohammed Y.H. Farooqui: Comparative toxicities of aliphatic nitriles. In: Toxicology Letters. Band 12, Nr. 2-3, Juli 1982, S. 157–163, doi:10.1016/0378-4274(82)90179-5.
- ↑ Kumbukani K. Nyirenda: Toxicity Potential of Cyanogenic Glycosides in Edible Plants. In: Medical Toxicology. IntechOpen, 3. Februar 2021.
- ↑ a b c d Fraser F. Fleming, Fraser F. Fleming: Nitrile-containing natural products. In: Natural Product Reports. Band 16, Nr. 5, 1999, S. 597–606, doi:10.1039/a804370a.
- ↑ a b c d Camille Scotti, James W. Barlow: Natural Products Containing the Nitrile Functional Group and Their Biological Activities. In: Natural Product Communications. Band 17, Nr. 5, Mai 2022, S. 1934578X2210999, doi:10.1177/1934578X221099973.
- ↑ Anete C Ferraz, Miriam Elizabeth M Angelucci, Mariana L Da Costa, Ilza R Batista, Bras H De Oliveira, Claudio Da Cunha: Pharmacological Evaluation of Ricinine, a Central Nervous System Stimulant Isolated from Ricinus communis. In: Pharmacology Biochemistry and Behavior. Band 63, Nr. 3, Juli 1999, S. 367–375, doi:10.1016/S0091-3057(99)00007-6.
- ↑ R. Mukherjee, A. Chatterjee: Structure and synthesis of nudiflorine. In: Tetrahedron. Band 22, Nr. 4, Januar 1966, S. 1461–1466, doi:10.1016/S0040-4020(01)99443-8.
- ↑ M.Soledade C Pedras, Corwin M Nycholat, Sabine Montaut, Yiming Xu, Abdul Q Khan: Chemical defenses of crucifers: elicitation and metabolism of phytoalexins and indole-3-acetonitrile in brown mustard and turnip. In: Phytochemistry. Band 59, Nr. 6, März 2002, S. 611–625, doi:10.1016/S0031-9422(02)00026-2.
- ↑ A. Bellirou, A. Bouali, B. Bouammali, N. Boukhatem, B.N. Elmtili, A. Hamal, M. El-Mourabit: Extraction of simmondsin and oil in one step from jojoba seeds. In: Industrial Crops and Products. Band 21, Nr. 2, März 2005, S. 229–233, doi:10.1016/j.indcrop.2004.04.007.
- ↑ Adolf Nahrstedt, Victor Wray: Structural revision of a putative cyanogenic glucoside from Ilex aquifolium. In: Phytochemistry. Band 29, Nr. 12, 1990, S. 3934–3936, doi:10.1016/0031-9422(90)85364-L.
- ↑ Wendy K. Swenson, John E. Dunn, Eric E. Conn: Cyanogenesis in Acacia sutherlandii. In: Phytochemistry. Band 26, Nr. 6, Januar 1987, S. 1835–1836, doi:10.1016/S0031-9422(00)82299-2.
- ↑ Martin G. Ettlinger, Jerzy W. Jaroszewski, Søren Rosendal Jensen, Bent Juhl Nielsen, Frederick Nartey: Proacacipetalin and acacipetalin. In: Journal of the Chemical Society, Chemical Communications. Nr. 24, 1977, S. 952, doi:10.1039/c39770000952.
- ↑ K Shanker, M Gupta, S Srivastava, D Bawankule, A Pal, S Khanuja: Determination of bioactive nitrile glycoside(s) in drumstick (Moringa oleifera) by reverse phase HPLC. In: Food Chemistry. Band 105, Nr. 1, 2007, S. 376–382, doi:10.1016/j.foodchem.2006.12.034.
- ↑ F. W. Stamler: Reproduction in Rats Fed Lathyrus Peas or Aminonitriles. In: Experimental Biology and Medicine. Band 90, Nr. 1, 1. Oktober 1955, S. 294–298, doi:10.3181/00379727-90-22013.
- ↑ E. D. Schilling, F. M. Strong: ISOLATION, STRUCTURE AND SYNTHESIS OF A LATHYRUS FACTOR FROM L. ODORATUS 1. In: Journal of the American Chemical Society. Band 76, Nr. 10, Mai 1954, S. 2848–2848, doi:10.1021/ja01639a084.
- ↑ Mohammadali Torbati, Hossein Nazemiyeh, Farzaneh Lotfipour, Solmaz Asnaashari, Mahboob Nemati, Fatemeh Fathiazad: Composition and Antibacterial Activity of Heracleum Transcaucasicum and Heracleum Anisactis Aerial Parts Essential Oil. In: Advanced Pharmaceutical Bulletin. 2013, doi:10.5681/APB.2013.066, PMID 24312869, PMC 3848220 (freier Volltext).
- ↑ Peter Lorenz, Sarina Duckstein, Jürgen Conrad, Matthias Knödler, Ulrich Meyer, Florian C. Stintzing: An Approach to the Chemotaxonomic Differentiation of Two European Dog's Mercury Species: Mercurialis annua L. and M. perennis L. In: Chemistry & Biodiversity. Band 9, Nr. 2, Februar 2012, S. 282–297, doi:10.1002/cbdv.201100341.
- ↑ K.L. Mikolajczak: Cyanolipids. In: Progress in the Chemistry of Fats and other Lipids. Band 15, Nr. 2, Januar 1977, S. 97–130, doi:10.1016/0079-6832(77)90013-1.
- ↑ David S. Seigler, Wanda Kawahara: New reports of cyanolipids from sapindaceous plants. In: Biochemical Systematics and Ecology. Band 4, Nr. 4, Januar 1976, S. 263–265, doi:10.1016/0305-1978(76)90050-8.
- ↑ E E Conn: Cyanogenic Compounds. In: Annual Review of Plant Physiology. Band 31, Nr. 1, Juni 1980, S. 433–451, doi:10.1146/annurev.pp.31.060180.002245.
- ↑ a b Pablo Díaz-Rueda, Laura Morales de los Ríos, Luis C Romero, Irene García: Old poisons, new signaling molecules: the case of hydrogen cyanide. In: Journal of Experimental Botany. Band 74, Nr. 19, 13. Oktober 2023, S. 6040–6051, doi:10.1093/jxb/erad317, PMID 37586035, PMC 10575699 (freier Volltext).
- ↑ Hieng-Ming Ting, Boon Huat Cheah, Yu-Cheng Chen, Pei-Min Yeh, Chiu-Ping Cheng, Freddy Kuok San Yeo, Ane Kjersti Vie, Jens Rohloff, Per Winge, Atle M. Bones, Ralph Kissen: The Role of a Glucosinolate-Derived Nitrile in Plant Immune Responses. In: Frontiers in Plant Science. Band 11, 10. März 2020, doi:10.3389/fpls.2020.00257, PMID 32211010, PMC 7076197 (freier Volltext).
- ↑ Adam M. Wentzell, Daniel J. Kliebenstein: Genotype, Age, Tissue, and Environment Regulate the Structural Outcome of Glucosinolate Activation. In: Plant Physiology. Band 147, Nr. 1, 28. April 2008, S. 415–428, doi:10.1104/pp.107.115279, PMID 18359845, PMC 2330308 (freier Volltext).
- ↑ Hideji Tanii: Allyl nitrile: Toxicity and health effects. In: Journal of Occupational Health. Band 59, Nr. 2, März 2017, S. 104–111, doi:10.1539/joh.16-0147-RA, PMID 28132970, PMC 5478528 (freier Volltext).
- ↑ Jinghua Yang, Zhangping Li, Jinmin Lian, Guoning Qi, Pibiao Shi, Jiawei He, Zhongyuan Hu, Mingfang Zhang: Brassicaceae transcriptomes reveal convergent evolution of super-accumulation of sinigrin. In: Communications Biology. Band 3, Nr. 1, 16. Dezember 2020, doi:10.1038/s42003-020-01523-x, PMID 33328568, PMC 7745032 (freier Volltext).
- ↑ David J. Williams, Christa Critchley, Sharon Pun, Mridusmita Chaliha, Timothy J. O’Hare: Differing mechanisms of simple nitrile formation on glucosinolate degradation in Lepidium sativum and Nasturtium officinale seeds. In: Phytochemistry. Band 70, Nr. 11-12, Juli 2009, S. 1401–1409, doi:10.1016/j.phytochem.2009.07.035.
- ↑ Ani Radonić, Ivica Blažević, Josip Mastelić, Marina Zekić, Mirjana Skočibušić, Ana Maravić: Phytochemical Analysis and Antimicrobial Activity of Cardaria draba (L.) Desv . Volatiles. In: Chemistry & Biodiversity. Band 8, Nr. 6, Juni 2011, S. 1170–1181, doi:10.1002/cbdv.201000370.
- ↑ Islamiyat F. Bolarinwa, Caroline Orfila, Michael R.A. Morgan: Amygdalin content of seeds, kernels and food products commercially-available in the UK. In: Food Chemistry. Band 152, Juni 2014, S. 133–139, doi:10.1016/j.foodchem.2013.11.002.
- ↑ C.J Graham: Nonstructural carbohydrate and prunasin composition of peach seedlings fertilized with different nitrogen sources and aluminum. In: Scientia Horticulturae. Band 94, Nr. 1-2, Mai 2002, S. 21–32, doi:10.1016/S0304-4238(01)00345-4.
- ↑ C.J Graham: Nonstructural carbohydrate and prunasin composition of peach seedlings fertilized with different nitrogen sources and aluminum. In: Scientia Horticulturae. Band 94, Nr. 1-2, Mai 2002, S. 21–32, doi:10.1016/S0304-4238(01)00345-4.
- ↑ Jandirk Sendker, Therese Ellendorff, Aljoscha Hölzenbein: Occurrence of Benzoic Acid Esters as Putative Catabolites of Prunasin in Senescent Leaves of Prunus laurocerasus. In: Journal of Natural Products. Band 79, Nr. 7, 22. Juli 2016, S. 1724–1729, doi:10.1021/acs.jnatprod.5b01090.
- ↑ David Chassagne, Jean C. Crouzet, Claude L. Bayonove, Raymond L. Baumes: Identification and Quantification of Passion Fruit Cyanogenic Glycosides. In: Journal of Agricultural and Food Chemistry. Band 44, Nr. 12, 1. Januar 1996, S. 3817–3820, doi:10.1021/jf960381t.
- ↑ David S. Seigler, Guido F. Pauli, Adolf Nahrstedt, Rosemary Leen: Cyanogenic allosides and glucosides from Passiflora edulis and Carica papaya. In: Phytochemistry. Band 60, Nr. 8, August 2002, S. 873–882, doi:10.1016/S0031-9422(02)00170-X.
- ↑ Mateja Senica, Franci Stampar, Robert Veberic, Maja Mikulic‐Petkovsek: The higher the better? Differences in phenolics and cyanogenic glycosides in Sambucus nigra leaves, flowers and berries from different altitudes. In: Journal of the Science of Food and Agriculture. Band 97, Nr. 8, Juni 2017, S. 2623–2632, doi:10.1002/jsfa.8085.
- ↑ Rex A. Buhrmester, John E. Ebinger, David S. Seigler: Sambunigrin and cyanogenic variability in populations of Sambucus canadensis L. (Caprifoliaceae). In: Biochemical Systematics and Ecology. Band 28, Nr. 7, August 2000, S. 689–695, doi:10.1016/S0305-1978(99)00105-2.
- ↑ Nhat Hao Tran Le, Karl Egil Malterud, Drissa Diallo, Berit Smestad Paulsen, Cecilie Sogn Nergård, Helle Wangensteen: Bioactive polyphenols in Ximenia americana and the traditional use among Malian healers. In: Journal of Ethnopharmacology. Band 139, Nr. 3, Februar 2012, S. 858–862, doi:10.1016/j.jep.2011.12.031.
- ↑ H. Kofod, R. Eyjólfsson: Cyanogenesis in species of the fern genera Cystopteris and Davalla. In: Phytochemistry. Band 8, Nr. 8, August 1969, S. 1509–1511, doi:10.1016/S0031-9422(00)85922-1.
- ↑ Gilles F. Nicollier, Daniel F. Pope, Alonzo C. Thompson: Biological activity of dhurrin and other compounds from Johnson grass (Sorghum halepense). In: Journal of Agricultural and Food Chemistry. Band 31, Nr. 4, Juli 1983, S. 744–748, doi:10.1021/jf00118a016.
- ↑ Tomas Laursen, Jonas Borch, Camilla Knudsen, Krutika Bavishi, Federico Torta, Helle Juel Martens, Daniele Silvestro, Nikos S. Hatzakis, Markus R. Wenk, Timothy R. Dafforn, Carl Erik Olsen, Mohammed Saddik Motawia, Björn Hamberger, Birger Lindberg Møller, Jean-Etienne Bassard: Characterization of a dynamic metabolon producing the defense compound dhurrin in sorghum. In: Science. Band 354, Nr. 6314, 18. November 2016, S. 890–893, doi:10.1126/science.aag2347.
- ↑ G.W. Butler: The distribution of the cyanoglucosides linamarin and lotaustralin in higher plants. In: Phytochemistry. Band 4, Nr. 1, Februar 1965, S. 127–131, doi:10.1016/S0031-9422(00)86154-3.
- ↑ M. P. Cereda, M.C.Y. Mattos: LINAMARIN: THE TOXIC COMPOUND OF CASSAVA. In: Journal of Venomous Animals and Toxins. Band 2, Nr. 1, 1996, S. 06–12, doi:10.1590/S0104-79301996000100002.
- ↑ Matthias Onyebuchi Agbo, Daowan Lai, Festus B.C. Okoye, Patience O. Osadebe, Peter Proksch: Antioxidative polyphenols from Nigerian mistletoe Loranthus micranthus (Linn.) parasitizing on Hevea brasiliensis. In: Fitoterapia. Band 86, April 2013, S. 78–83, doi:10.1016/j.fitote.2013.02.006.
- ↑ R. Lieberei, D. Selmar, B. Biehl: Metabolization of cyanogenic glucosides in Hevea brasiliensis. In: Plant Systematics and Evolution. Band 150, Nr. 1-2, 1985, S. 49–63, doi:10.1007/BF00985567.
- ↑ a b Mika Zagrobelny, Érika de Castro, Birger Møller, Søren Bak: Cyanogenesis in Arthropods: From Chemical Warfare to Nuptial Gifts. In: Insects. Band 9, Nr. 2, 3. Mai 2018, S. 51, doi:10.3390/insects9020051, PMID 29751568, PMC 6023451 (freier Volltext).
- ↑ Ritsuo Nishida, Miriam Rothschild, Rosemary Mummery: Acyanoglucoside, sarmentosin, from the magpie moth, Abraxas grossulariata, geometridae: Lepidoptera. In: Phytochemistry. Band 36, Nr. 1, Mai 1994, S. 37–38, doi:10.1016/S0031-9422(00)97007-9.
- ↑ Nanna Bjarnholt, Mirosław Nakonieczny, Andrzej Kędziorski, Diane M. Debinski, Stephen F. Matter, Carl Erik Olsen, Mika Zagrobelny: Occurrence of Sarmentosin and Other Hydroxynitrile Glucosides in Parnassius (Papilionidae) Butterflies and Their Food Plants. In: Journal of Chemical Ecology. Band 38, Nr. 5, Mai 2012, S. 525–537, doi:10.1007/s10886-012-0114-x.
- ↑ J. R. Aldrich, S. P. Carroll, W. R. Lusby, M. J. Thompson, J. P. Kochansky, R. M. Waters: Sapindaceae, cyanolipids, and bugs. In: Journal of Chemical Ecology. Band 16, Nr. 1, Januar 1990, S. 199–210, doi:10.1007/BF01021279.
- ↑ J.C. Braekman, D. Daloze, J.M. Pasteels: Cyanogenic and other glucosides in a neo-guinean bug Leptocoris isolata: Possible precursors in its host-plant. In: Biochemical Systematics and Ecology. Band 10, Nr. 4, Dezember 1982, S. 355–364, doi:10.1016/0305-1978(82)90010-2.
- ↑ Mika Zagrobelny, Karsten Scheibye-Alsing, Niels Bjerg Jensen, Birger Lindberg Møller, Jan Gorodkin, Søren Bak: 454 pyrosequencing based transcriptome analysis of Zygaena filipendulae with focus on genes involved in biosynthesis of cyanogenic glucosides. In: BMC Genomics. Band 10, Nr. 1, Dezember 2009, doi:10.1186/1471-2164-10-574, PMID 19954531, PMC 2791780 (freier Volltext).
- ↑ Adolf Nahrstedt: Cyanogenesis and foodplants. In: Phytochemistry and Agriculture. Oxford University PressOxford, 1993, ISBN 978-0-19-857762-1, S. 107–129.
- ↑ Ljubodrag V. Vujisić, Ivan M. Vučković, Slobodan E. Makarov, Bojan S. Ilić, Dragan Ž. Antić, Milka B. Jadranin, Nina M. Todorović, Ivan V. Mrkić, Vlatka E. Vajs, Luka R. Lučić, Božidar P. M. Ćurčić, Bojan M. Mitić: Chemistry of the sternal gland secretion of the Mediterranean centipede Himantarium gabrielis (Linnaeus, 1767) (Chilopoda: Geophilomorpha: Himantariidae). In: Naturwissenschaften. Band 100, Nr. 9, September 2013, S. 861–870, doi:10.1007/s00114-013-1086-6.
- ↑ Karsten Seidelmann, Heike Weinert, Hans-Jörg Ferenz: Wings and legs are production sites for the desert locust courtship-inhibition pheromone, phenylacetonitrile. In: Journal of Insect Physiology. Band 49, Nr. 12, Dezember 2003, S. 1125–1133, doi:10.1016/j.jinsphys.2003.08.005.
- ↑ S. S. Duffey, M. S. Blum, H. M. Fales, S. L. Evans, R. W. Roncadori, D. L. Tiemann, Y. Nakagawa: Benzoyl cyanide and mandelonitrile benzoate in the defensive secretions of millipedes. In: Journal of Chemical Ecology. Band 3, Nr. 1, 1977, S. 101–113, doi:10.1007/BF00988137.
- ↑ Adrian Brückner, Günther Raspotnig, Katja Wehner, Reinhard Meusinger, Roy A. Norton, Michael Heethoff: Storage and release of hydrogen cyanide in a chelicerate ( Oribatula tibialis ). In: Proceedings of the National Academy of Sciences. Band 114, Nr. 13, 28. März 2017, S. 3469–3472, doi:10.1073/pnas.1618327114, PMID 28289203, PMC 5380029 (freier Volltext).
- ↑ Guido Cimino, Margherita Gavagnin, Guido Sodano, Aldo Spinella, Giuseppe Strazzullo, Francis J. Schmitz, Gopichand Yalamanchili: Revised structure of bursatellin. In: The Journal of Organic Chemistry. Band 52, Nr. 11, Mai 1987, S. 2301–2303, doi:10.1021/jo00387a037.
- ↑ Annika Fagerholm, Damien Habrant, Ari M. Koskinen: Calyculins and Related Marine Natural Products as Serine-Threonine Protein Phosphatase PP1 and PP2A Inhibitors and Total Syntheses of Calyculin A, B, and C. In: Marine Drugs. Band 8, Nr. 1, 21. Januar 2010, S. 122–172, doi:10.3390/md80100122, PMID 20161975, PMC 2817927 (freier Volltext).
- ↑ Samuele Sala, Jane Fromont, Oliver Gomez, Daniel Vuong, Ernest Lacey, Gavin R. Flematti: Albanitriles A–G: Antiprotozoal Polyacetylene Nitriles from a Mycale Marine Sponge. In: Journal of Natural Products. Band 82, Nr. 12, 27. Dezember 2019, S. 3450–3455, doi:10.1021/acs.jnatprod.9b00840.
- ↑ C J Knowles: Microorganisms and cyanide. In: Bacteriological Reviews. Band 40, Nr. 3, September 1976, S. 652–680, doi:10.1128/br.40.3.652-680.1976, PMID 791236, PMC 413975 (freier Volltext).
- ↑ Hideyuki Takahashi, Koohei Nozawa, Ken-ichi Kawai: Isolation and Structures of Dicyanide Derivatives, Epurpurins A to C, from Emericella purpurea. In: Chemical and Pharmaceutical Bulletin. Band 44, Nr. 12, 1996, S. 2227–2230, doi:10.1248/cpb.44.2227.
- ↑ Marjorie Anchel: Metabolic products of Clitocybe diatreta. I. Diatretyne amide and diatretyne nitrile. In: Archives of Biochemistry and Biophysics. Band 78, Nr. 1, November 1958, S. 100–110, doi:10.1016/0003-9861(58)90318-7.
- ↑ N. G. Heatley, J. S. Stephenson: Identity of ‘Nudic Acid B’ and ‘Diatretyne II’. In: Nature. Band 179, Nr. 4569, Mai 1957, S. 1078–1078, doi:10.1038/1791078a0.
- ↑ Jan Caspar, Peter Spiteller: A Free Cyanohydrin as Arms and Armour of Marasmius oreades. In: ChemBioChem. Band 16, Nr. 4, 2. März 2015, S. 570–573, doi:10.1002/cbic.201402453.
- ↑ Anju Sehrawat, Satyavir S. Sindhu, Bernard R. Glick: Hydrogen cyanide production by soil bacteria: Biological control of pests and promotion of plant growth in sustainable agriculture. In: Pedosphere. Band 32, Nr. 1, Februar 2022, S. 15–38, doi:10.1016/S1002-0160(21)60058-9.
- ↑ Diogo Montes Vidal, Anna‐Lena von Rymon‐Lipinski, Srinivasa Ravella, Ulrike Groenhagen, Jennifer Herrmann, Nestor Zaburannyi, Paulo H. G. Zarbin, Adithi R. Varadarajan, Christian H. Ahrens, Laure Weisskopf, Rolf Müller, Stefan Schulz: Long‐Chain Alkyl Cyanides: Unprecedented Volatile Compounds Released by Pseudomonas and Micromonospora Bacteria. In: Angewandte Chemie International Edition. Band 56, Nr. 15, 3. April 2017, S. 4342–4346, doi:10.1002/anie.201611940.
- ↑ Joel P. Cioni, James R. Doroghazi, Kou-San Ju, Xiaomin Yu, Bradley S. Evans, Jaeheon Lee, William W. Metcalf: Cyanohydrin Phosphonate Natural Product from Streptomyces regensis. In: Journal of Natural Products. Band 77, Nr. 2, 28. Februar 2014, S. 243–249, doi:10.1021/np400722m, PMID 24437999, PMC 3993929 (freier Volltext).
- ↑ Sanjoy Adak, April L. Lukowski, Rebecca J. B. Schäfer, Bradley S. Moore: From Tryptophan to Toxin: Nature’s Convergent Biosynthetic Strategy to Aetokthonotoxin. In: Journal of the American Chemical Society. Band 144, Nr. 7, 23. Februar 2022, S. 2861–2866, doi:10.1021/jacs.1c12778, PMID 35142504, PMC 9004672 (freier Volltext).
- ↑ a b Max P. Bernstein, Samantha F. M. Ashbourn, Scott A. Sandford, Louis J. Allamandola: The Lifetimes of Nitriles (CN) and Acids (COOH) during Ultraviolet Photolysis and Their Survival in Space. In: The Astrophysical Journal. Band 601, Nr. 1, 20. Januar 2004, S. 365–370, doi:10.1086/380306.
- ↑ S Green: Interstellar Chemistry: Exotic Molecules in Space. In: Annual Review of Physical Chemistry. Band 32, Nr. 1, Oktober 1981, S. 103–138, doi:10.1146/annurev.pc.32.100181.000535.
- ↑ Richard Carter, Mary M. Nijhout: Control of Gamete Formation (Exflagellation) in Malaria Parasites. In: Science. Band 195, Nr. 4276, 28. Januar 1977, S. 407–409, doi:10.1126/science.12566.
- ↑ Jean-Claude Guillemin, Miloud Bouyahyi, El Hassan Riague: Prebiotic, planetary and interstellar chemistry starting from compounds detected in the interstellar medium. In: Advances in Space Research. Band 33, Nr. 1, Januar 2004, S. 81–87, doi:10.1016/j.asr.2003.07.015.
- ↑ a b c Nicholas F. Wogan, David C. Catling, Kevin J. Zahnle, Roxana Lupu: Origin-of-life Molecules in the Atmosphere after Big Impacts on the Early Earth. In: The Planetary Science Journal. Band 4, Nr. 9, 1. September 2023, S. 169, doi:10.3847/PSJ/aced83.
- ↑ a b Yannick Vallee, Ibrahim Shalayel, Kieu-Dung Ly, K. V. Raghavendra Rao, Gael De Paëpe, Katharina Märker, Anne Milet: At the very beginning of life on Earth: the thiol-rich peptide (TRP) world hypothesis. In: The International Journal of Developmental Biology. Band 61, Nr. 8-9, 2017, S. 471–478, doi:10.1387/ijdb.170028yv.
- ↑ a b James P. Ferris, William J. Hagan: HCN and chemical evolution: The possible role of cyano compounds in prebiotic synthesis. In: Tetrahedron. Band 40, Nr. 7, Januar 1984, S. 1093–1120, doi:10.1016/S0040-4020(01)99315-9.
- ↑ L. Chimiak, J. Eiler, A. Sessions, C. Blumenfeld, M. Klatte, B.M. Stoltz: Isotope effects at the origin of life: Fingerprints of the Strecker synthesis. In: Geochimica et Cosmochimica Acta. Band 321, März 2022, S. 78–98, doi:10.1016/j.gca.2022.01.015.
- ↑ Ibrahim Shalayel, Seydou Coulibaly, Kieu Ly, Anne Milet, Yannick Vallée: The Reaction of Aminonitriles with Aminothiols: A Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean. In: Life. Band 8, Nr. 4, 19. Oktober 2018, S. 47, doi:10.3390/life8040047, PMID 30347745, PMC 6316830 (freier Volltext).
- ↑ a b Thomas Laue, Andreas Plagens: Namen- und Schlagwort-Reaktionen der Organischen Chemie. In: Teubner Studienbücher Chemie. 1994, doi:10.1007/978-3-322-94726-0.
- ↑ K. R. Lynn, Peter E. Yankwich: Cyanide Carbon Isotope Fractionation in the Reaction of Cyanide Ion and Methyl Iodide. Carbon Isotope Effect in the Hydrolysis of Methyl Iodide. In: Journal of the American Chemical Society. Band 83, Nr. 1, Januar 1961, S. 53–57, doi:10.1021/ja01462a010.
- ↑ G. E. Ham, Jane Stevens: Reaction of 1,2-Dihaloethanes with Sodium Cyanide. In: The Journal of Organic Chemistry. Band 27, Nr. 12, Dezember 1962, S. 4638–4639, doi:10.1021/jo01059a504.
- ↑ Cinzia Chiappe, Daniela Pieraccini, Paola Saullo: Nucleophilic Displacement Reactions in Ionic Liquids: Substrate and Solvent Effect in the Reaction of NaN 3 and KCN with Alkyl Halides and Tosylates. In: The Journal of Organic Chemistry. Band 68, Nr. 17, 1. August 2003, S. 6710–6715, doi:10.1021/jo026838h.
- ↑ W. Nagata, M. Yoshioka, S. Hirai: Hydrocyanation. IV. New hydrocyanation methods using hydrogen cyanide and an alkylaluminum, and an alkylaluminum cyanide. In: Journal of the American Chemical Society. Band 94, Nr. 13, Juni 1972, S. 4635–4643, doi:10.1021/ja00768a037.
- ↑ W. Nagata, M. Yoshioka, T. Okumura: Hydrocyanation. Part X. Cleavage of epoxides with hydrogen cyanide and triethylaluminium and with diethylaluminium cyanide. In: Journal of the Chemical Society C: Organic. Nr. 17, 1970, S. 2365, doi:10.1039/j39700002365.
- ↑ Jeffrey C. Mullis, William P. Weber: Regiospecificity of reactions of epoxides and oxetanes with trimethylsilyl cyanide. In: The Journal of Organic Chemistry. Band 47, Nr. 15, Juli 1982, S. 2873–2875, doi:10.1021/jo00136a011.
- ↑ Hongru Zhang, Xin Su, Kaiwu Dong: Recent progress in transition-metal-catalyzed hydrocyanation of nonpolar alkenes and alkynes. In: Organic & Biomolecular Chemistry. Band 18, Nr. 3, 2020, S. 391–399, doi:10.1039/C9OB02374G.
- ↑ a b Muthupandian Ganesan, Paramathevar Nagaraaj: Recent developments in dehydration of primary amides to nitriles. In: Organic Chemistry Frontiers. Band 7, Nr. 22, 2020, S. 3792–3814, doi:10.1039/D0QO00843E.
- ↑ a b Dilip Konwar, Monalisa Boruah, Gautom Kumar Sarmah, Nayan Kamal Bhattacharyya, Naleen Borthakur, Birendra Nath Goswami, Kumar Ranjan Boruah: Aluminium Chloride and Sodium Iodide (AlCl3-NaI): A Versatile Dehydrating Agent. In: Journal of Chemical Research. Band 2001, Nr. 11, November 2001, S. 490–492, doi:10.3184/030823401103168604.
- ↑ a b Imen Talbi, Mohamed Lotfi Efrit, Soufiane Touil: Efficient New Protocols for Converting Primary Amides into Nitriles Initiated by P(NMe 2 ) 3 , PCl 3 , or P(OPh) 3. In: ACS Omega. Band 3, Nr. 5, 31. Mai 2018, S. 5078–5082, doi:10.1021/acsomega.8b00544, PMID 31458722, PMC 6641971 (freier Volltext).
- ↑ Muthupandian Ganesan: Methods for Direct Conversion of Primary Nitroalkanes to Nitriles. In: Current Organic Chemistry. Band 25, Nr. 24, 22. Dezember 2021, S. 2990–3003, doi:10.2174/1385272825666211126124835.
- ↑ Z. Shahsavari-Fard, A. R. Sardarian: Diethyl chlorophosphate: A new alternative reagent for dehydration of primary amides to nitriles in solvent and solvent-free conditions. In: Journal of the Iranian Chemical Society. Band 8, Nr. 1, März 2011, S. 204–208, doi:10.1007/BF03246217.
- ↑ 2-ETHYLHEXANONITRILE. In: Organic Syntheses. Band 32, 1952, S. 65, doi:10.15227/orgsyn.032.0065.
- ↑ Harry Babad, Andrew G. Zeiler: Chemistry of phosgene. In: Chemical Reviews. Band 73, Nr. 1, 1. Februar 1973, S. 75–91, doi:10.1021/cr60281a005.
- ↑ Mohammed H. Al‐Huniti, Mitchell P. Croatt: Metal‐Catalyzed Dehydration of Primary Amides to Nitriles. In: Asian Journal of Organic Chemistry. Band 8, Nr. 10, Oktober 2019, S. 1791–1799, doi:10.1002/ajoc.201900343.
- ↑ Hiroyuki Okabe, Asuka Naraoka, Takahiro Isogawa, Shunsuke Oishi, Hiroshi Naka: Acceptor-Controlled Transfer Dehydration of Amides to Nitriles. In: Organic Letters. Band 21, Nr. 12, 21. Juni 2019, S. 4767–4770, doi:10.1021/acs.orglett.9b01657.
- ↑ Stephan Enthaler: Straightforward Iron‐Catalyzed Synthesis of Nitriles by Dehydration of Primary Amides. In: European Journal of Organic Chemistry. Band 2011, Nr. 25, September 2011, S. 4760–4763, doi:10.1002/ejoc.201100754.
- ↑ Stephan Enthaler: Straightforward Uranium‐Catalyzed Dehydration of Primary Amides to Nitriles. In: Chemistry – A European Journal. Band 17, Nr. 34, 16. August 2011, S. 9316–9319, doi:10.1002/chem.201101478.
- ↑ Stephan Enthaler, Shigeyoshi Inoue: An Efficient Zinc‐Catalyzed Dehydration of Primary Amides to Nitriles. In: Chemistry – An Asian Journal. Band 7, Nr. 1, 2. Januar 2012, S. 169–175, doi:10.1002/asia.201100493.
- ↑ Shekharappa, L. Roopesh Kumar, C. Srinivasulu, Vommina V. Sureshbabu: Dehydration of Chiral α-Amides to Chiral α-Nitriles Under the Appel Reaction Conditions. In: International Journal of Peptide Research and Therapeutics. Band 27, Nr. 1, März 2021, S. 497–502, doi:10.1007/s10989-020-10101-y.
- ↑ Richard S. Monson, Deggary N. Priest: Dehydration of Amides to Nitriles Initiated by Hexamethylphosphoric Triamide. In: Canadian Journal of Chemistry. Band 49, Nr. 17, 1. September 1971, S. 2897–2898, doi:10.1139/v71-480.
- ↑ Krishnappa Manjula, Mohamed Afzal Pasha: Rapid Method of Converting Primary Amides to Nitriles and Nitriles to Primary Amides by ZnCl 2 using Microwaves under Different Reaction Conditions. In: Synthetic Communications. Band 37, Nr. 9, Mai 2007, S. 1545–1550, doi:10.1080/00397910701230147.
- ↑ Rui Ding, Yongguo Liu, Mengru Han, Wenyi Jiao, Jiaqi Li, Hongyu Tian, Baoguo Sun: Synthesis of Nitriles from Primary Amides or Aldoximes under Conditions of a Catalytic Swern Oxidation. In: The Journal of Organic Chemistry. Band 83, Nr. 20, 19. Oktober 2018, S. 12939–12944, doi:10.1021/acs.joc.8b02190.
- ↑ Kazuaki Ishihara, Yoshiro Furuya, Hisashi Yamamoto: Rhenium(VII) Oxo Complexes as Extremely Active Catalysts in the Dehydration of Primary Amides and Aldoximes to Nitriles. In: Angewandte Chemie. Band 114, Nr. 16, 16. August 2002, S. 3109, doi:10.1002/1521-3757(20020816)114:16<3109::AID-ANGE3109>3.0.CO;2-K.
- ↑ Jiban K. Chakrabarti, Terrence M. Hotten: A new route to nitriles. Dehydration of aldoximes using 2,4,6-trichloro-s-triazine (cyanuric chloride). In: Journal of the Chemical Society, Chemical Communications. Nr. 22, 1972, S. 1226, doi:10.1039/c39720001226.
- ↑ Mild and Efficient Dehydration of Oximes to Nitriles Mediated by the Burgess Reagent. In: Synlett. Band 2000, Nr. 08, 2000, S. 1169–1171, doi:10.1055/s-2000-6752.
- ↑ Ziad Moussa, Saleh A. Ahmed, Ahmad S. ElDouhaibi, Shaya Y. Al-Raqa: NMR Studies and electrophilic properties of triphenylphosphine–trifluoromethanesulfonic anhydride; a remarkable dehydrating reagent system for the conversion of aldoximes into nitriles. In: Tetrahedron Letters. Band 51, Nr. 14, April 2010, S. 1826–1831, doi:10.1016/j.tetlet.2010.01.119.
- ↑ Kengo Hyodo, Saki Kitagawa, Masayuki Yamazaki, Kingo Uchida: Iron‐Catalyzed Dehydration of Aldoximes to Nitriles Requiring Neither Other Reagents Nor Nitrile Media. In: Chemistry – An Asian Journal. Band 11, Nr. 9, 6. Mai 2016, S. 1348–1352, doi:10.1002/asia.201600085.
- ↑ Philipp Rommelmann, Tobias Betke, Harald Gröger: Synthesis of Enantiomerically Pure N -Acyl Amino Nitriles via Catalytic Dehydration of Oximes and Application in a de Novo Synthesis of Vildagliptin. In: Organic Process Research & Development. Band 21, Nr. 10, 20. Oktober 2017, S. 1521–1527, doi:10.1021/acs.oprd.7b00169.
- ↑ a b Kazuya Yamaguchi, Hiroshi Fujiwara, Yoshiyuki Ogasawara, Miyuki Kotani, Noritaka Mizuno: A Tungsten–Tin Mixed Hydroxide as an Efficient Heterogeneous Catalyst for Dehydration of Aldoximes to Nitriles. In: Angewandte Chemie. Band 119, Nr. 21, 18. Mai 2007, S. 3996–3999, doi:10.1002/ange.200605004.
- ↑ Dongliang Zhang, Yaping Huang, Erlei Zhang, Rong Yi, Chao Chen, Lei Yu, Qing Xu: Pd/Mn Bimetallic Relay Catalysis for Aerobic Aldoxime Dehydration to Nitriles. In: Advanced Synthesis & Catalysis. Band 360, Nr. 4, 15. Februar 2018, S. 784–790, doi:10.1002/adsc.201701154.
- ↑ Tobias Betke, Jun Higuchi, Philipp Rommelmann, Keiko Oike, Taiji Nomura, Yasuo Kato, Yasuhisa Asano, Harald Gröger: Biocatalytic Synthesis of Nitriles through Dehydration of Aldoximes: The Substrate Scope of Aldoxime Dehydratases. In: ChemBioChem. Band 19, Nr. 8, 16. April 2018, S. 768–779, doi:10.1002/cbic.201700571.
- ↑ Liyuan Lan, Shuai Huang, Yongguo Liu, Baoguo Sun, Hongyu Tian: Preparation and odor characteristics of nitriles derived from aldehydes. In: Flavour and Fragrance Journal. Band 35, Nr. 4, Juli 2020, S. 425–434, doi:10.1002/ffj.3581.
- ↑ Dylan J. Quinn, Graham J. Haun, Gustavo Moura-Letts: Direct synthesis of nitriles from aldehydes with hydroxylamine-O-sulfonic acid in acidic water. In: Tetrahedron Letters. Band 57, Nr. 34, August 2016, S. 3844–3847, doi:10.1016/j.tetlet.2016.07.047.
- ↑ Xiao-De An, Shouyun Yu: Direct Synthesis of Nitriles from Aldehydes Using an O -Benzoyl Hydroxylamine (BHA) as the Nitrogen Source. In: Organic Letters. Band 17, Nr. 20, 16. Oktober 2015, S. 5064–5067, doi:10.1021/acs.orglett.5b02547.
- ↑ Antonella Leggio, Emilia Lucia Belsito, Sonia Gallo, Angelo Liguori: One-pot conversion of aldehydes to nitriles mediated by TiCl 4. In: Tetrahedron Letters. Band 58, Nr. 15, April 2017, S. 1512–1514, doi:10.1016/j.tetlet.2017.03.007.
- ↑ Lewis Acid Reagents Edited by H. Yamamoto. Oxford University Press: Oxford, UK. 1999. 270 pp. £75.00, ISBN 0 19 850099 8. In: Organic Process Research & Development. Band 3, Nr. 4, 15. April 1999, S. 292–292, doi:10.1021/op990022+.
- ↑ Jitendra Gurjar, Jorick Bater, Valery V. Fokin: Sulfuryl Fluoride Mediated Conversion of Aldehydes to Nitriles. In: Chemistry – A European Journal. Band 25, Nr. 8, 6. Februar 2019, S. 1906–1909, doi:10.1002/chem.201805175.
- ↑ Albert M. van Leusen, Piet G. Oomkes: One-Step Conversion of Aldehydes to Nitriles. Introduction of a One-Carbon Unit. In: Synthetic Communications. Band 10, Nr. 5, Januar 1980, S. 399–403, doi:10.1080/00397918008061830.
- ↑ Otto H. Oldenziel, Daan Van Leusen, Albert M. Van Leusen: Chemistry of sulfonylmethyl isocyanides. 13. A general one-step synthesis of nitriles from ketones using tosylmethyl isocyanide. Introduction of a one-carbon unit. In: The Journal of Organic Chemistry. Band 42, Nr. 19, September 1977, S. 3114–3118, doi:10.1021/jo00439a002.
- ↑ Niamh Disney, Megan Smyth, Scott Wharry, Thomas S. Moody, Marcus Baumann: A cyanide-free synthesis of nitriles exploiting flow chemistry. In: Reaction Chemistry & Engineering. 2024, doi:10.1039/D3RE00458A.
- ↑ M. F. Semmelhack, Christopher R. Schmid: Nitroxyl-mediated electro-oxidation of amines to nitriles and carbonyl compounds. In: Journal of the American Chemical Society. Band 105, Nr. 22, Oktober 1983, S. 6732–6734, doi:10.1021/ja00360a042.
- ↑ Kyle M. Lambert, James M. Bobbitt, Sherif A. Eldirany, Liam E. Kissane, Rose K. Sheridan, Zachary D. Stempel, Francis H. Sternberg, William F. Bailey: Metal‐Free Oxidation of Primary Amines to Nitriles through Coupled Catalytic Cycles. In: Chemistry – A European Journal. Band 22, Nr. 15, 4. April 2016, S. 5156–5159, doi:10.1002/chem.201600549.
- ↑ Yasunari Maeda, Takahiro Nishimura, Sakae Uemura: Copper-Catalyzed Oxidation of Amines with Molecular Oxygen. In: Bulletin of the Chemical Society of Japan. Band 76, Nr. 12, Dezember 2003, S. 2399–2403, doi:10.1246/bcsj.76.2399.
- ↑ Christiane Janke, Jörg Radnik, Ursula Bentrup, Andreas Martin, Angelika Brückner: Vanadium‐Containing Oxynitrides: Effective Catalysts for the Ammoxidation of 3‐Picoline. In: ChemCatChem. Band 1, Nr. 4, 30. November 2009, S. 485–491, doi:10.1002/cctc.200900180.
- ↑ a b c David Nakles, Richard Luthy, George Wong-Chong: Manufacture and the Use of Cyanide. In: Cyanide in Water and Soil. CRC Press, 9. Dezember 2005, S. 41–55.
- ↑ Yoshiaki Nakao, Tamejiro Hiyama: Nickel-catalyzed carbocyanation of alkynes. In: Pure and Applied Chemistry. Band 80, Nr. 5, 1. Januar 2008, S. 1097–1107, doi:10.1351/pac200880051097.
- ↑ Haitham Hassan, Vincent Pirenne, Maren Wissing, Chahinaz Khiar, Ashique Hussain, Frédéric Robert, Yannick Landais: Free‐Radical Carbocyanation of Olefins. In: Chemistry – A European Journal. Band 23, Nr. 19, 3. April 2017, S. 4651–4658, doi:10.1002/chem.201605946.
- ↑ Laurent Vanoye, Ahmad Hammoud, Hélène Gérard, Alexandra Barnes, Régis Philippe, Pascal Fongarland, Claude de Bellefon, Alain Favre-Réguillon: Direct Synthesis of Nitriles from Carboxylic Acids Using Indium-Catalyzed Transnitrilation: Mechanistic and Kinetic Study. In: ACS Catalysis. Band 9, Nr. 11, 1. November 2019, S. 9705–9714, doi:10.1021/acscatal.9b02779.
- ↑ Tetsuto Tsunoda, Kaori Uemoto, Chisato Nagino, Megumi Kawamura, Hiroto Kaku, Shô Itô: A facile one-pot cyanation of primary and secondary alcohols. Application of some new Mitsunobu reagents. In: Tetrahedron Letters. Band 40, Nr. 41, Oktober 1999, S. 7355–7358, doi:10.1016/S0040-4039(99)01509-9.
- ↑ Walter-Georg Veeck, Manfred Regitz: Triple-bonded Heteroatom Derivatives Other Than Nitriles with Another Heteroatom Attached to the sp-Carbon Atom. In: Comprehensive Organic Functional Group Transformations. Elsevier, 1995, S. 1151–1160.
- ↑ B.A. Phillips, G. Fodor, J. Gal, F. Letourneau, J.J. Ryan: Mechanism of the von Braun amide degradations with carbonyl bromide or phosphorus pentabromide. In: Tetrahedron. Band 29, Nr. 21, Januar 1973, S. 3309–3327, doi:10.1016/S0040-4020(01)93483-0.
- ↑ Irina P. Beletskaya, Alexander S. Sigeev, Alexander S. Peregudov, Pavel V. Petrovskii: Catalytic Sandmeyer cyanation as a synthetic pathway to aryl nitriles. In: Journal of Organometallic Chemistry. Band 689, Nr. 23, November 2004, S. 3810–3812, doi:10.1016/j.jorganchem.2004.07.019.
- ↑ C. Frederick Koelsch: Some Applications of the Rosenmund-v. Braun Nitrile Synthesis. In: Journal of the American Chemical Society. Band 58, Nr. 8, August 1936, S. 1328–1330, doi:10.1021/ja01299a004.
- ↑ Naoto Chatani, Terukiyo Hanafusa: Transition-metal-catalyzed reactions of trimethylsilyl cyanide. 4. Palladium-catalyzed cyanation of aryl halides by trimethylsilyl cyanide. In: The Journal of Organic Chemistry. Band 51, Nr. 24, November 1986, S. 4714–4716, doi:10.1021/jo00374a041.
- ↑ Shunichi Murahashi, Takeshi Naota, Nobuyuki Nakajima: Palladium-catalyzed decarbonylation of acyl cyanides. In: The Journal of Organic Chemistry. Band 51, Nr. 6, März 1986, S. 898–901, doi:10.1021/jo00356a029.
- ↑ Mark Sundermeier, Alexander Zapf, Matthias Beller, Jürgen Sans: A new palladium catalyst system for the cyanation of aryl chlorides. In: Tetrahedron Letters. Band 42, Nr. 38, September 2001, S. 6707–6710, doi:10.1016/S0040-4039(01)01390-9.
- ↑ Todd D. Senecal, Wei Shu, Stephen L. Buchwald: A General, Practical Palladium‐Catalyzed Cyanation of (Hetero)Aryl Chlorides and Bromides. In: Angewandte Chemie. Band 125, Nr. 38, 16. September 2013, S. 10219–10223, doi:10.1002/ange.201304188.
- ↑ Florian Glöcklhofer, Markus Lunzer, Johannes Fröhlich: Facile Synthesis of Cyanoarenes from Quinones by Reductive Aromatization of Cyanohydrin Intermediates. In: Synlett. Band 26, Nr. 07, 1. April 2015, S. 950–952, doi:10.1055/s-0034-1380150.
- ↑ Jonathan T. Reeves, Christian A. Malapit, Frederic G. Buono, Kanwar P. Sidhu, Maurice A. Marsini, C. Avery Sader, Keith R. Fandrick, Carl A. Busacca, Chris H. Senanayake: Transnitrilation from Dimethylmalononitrile to Aryl Grignard and Lithium Reagents: A Practical Method for Aryl Nitrile Synthesis. In: Journal of the American Chemical Society. Band 137, Nr. 29, 29. Juli 2015, S. 9481–9488, doi:10.1021/jacs.5b06136.
- ↑ Hiroshi Ohno, Atsunori Mori, Shohei Inoue: Lanthanoid(III) Alkoxides as Novel Catalysts for a Rapid Transhydrocyanation from Acetone Cyanohydrin to Aldehydes and Ketones. In: Chemistry Letters. Band 22, Nr. 2, Februar 1993, S. 375–378, doi:10.1246/cl.1993.375.
- ↑ David A. Evans, Gary L. Carroll, Larry K. Truesdale: Synthetic applications of trimethylsilyl cyanide. Efficient synthesis of β-aminomethyl alcohols. In: The Journal of Organic Chemistry. Band 39, Nr. 7, April 1974, S. 914–917, doi:10.1021/jo00921a012.
- ↑ Shu Kobayashi, Yoshikazu Tsuchiya, Teruaki Mukaiyama: A Facile Synthesis of Cyanohydrin Trimethylsilyl Ethers by the Addition Reaction of Trimethylsilyl Cyanide with Aldehydes under Basic Condition. In: Chemistry Letters. Band 20, Nr. 4, April 1991, S. 537–540, doi:10.1246/cl.1991.537.
- ↑ Yuri N. Belokon', Susana Caveda-Cepas, Brendan Green, Nicolai S. Ikonnikov, Viktor N. Khrustalev, Vladimir S. Larichev, Margarita A. Moscalenko, Michael North, Charles Orizu, Vitali I. Tararov, Michela Tasinazzo, Galina I. Timofeeva, Lidia V. Yashkina: The Asymmetric Addition of Trimethylsilyl Cyanide to Aldehydes Catalyzed by Chiral (Salen)Titanium Complexes. In: Journal of the American Chemical Society. Band 121, Nr. 16, 1. April 1999, S. 3968–3973, doi:10.1021/ja984197v.
- ↑ a b Nobuhito Kurono, Takeshi Ohkuma: Catalytic Asymmetric Cyanation Reactions. In: ACS Catalysis. Band 6, Nr. 2, 5. Februar 2016, S. 989–1023, doi:10.1021/acscatal.5b02184.
- ↑ Siegfried Hünig, Rainer Schaller: The Chemistry of Acyl Cyanides. In: Angewandte Chemie International Edition in English. Band 21, Nr. 1, Januar 1982, S. 36–49, doi:10.1002/anie.198200361.
- ↑ Harald Gröger, Yasuhisa Asano: Cyanide-Free Enantioselective Catalytic Strategies for the Synthesis of Chiral Nitriles. In: The Journal of Organic Chemistry. doi:10.1021/acs.joc.9b02773.
- ↑ a b c d Amitava Rakshit, Hirendra Nath Dhara, Ashish Kumar Sahoo, Bhisma K. Patel: The Renaissance of Organo Nitriles in Organic Synthesis. In: Chemistry – An Asian Journal. Band 17, Nr. 21, 2. November 2022, doi:10.1002/asia.202200792.
- ↑ Charles R. Hauser, David S. Hoffenberg: CONVERSION OF NITRILES TO AMIDES AND ACIDS BY MEANS OF BORON FLUORIDE. In: The Journal of Organic Chemistry. Band 20, Nr. 10, Oktober 1955, S. 1448–1453, doi:10.1021/jo01127a025.
- ↑ V. G. Debabov, A. S. Yanenko: Biocatalytic hydrolysis of nitriles. In: Review Journal of Chemistry. Band 1, Nr. 4, Oktober 2011, S. 385–402, doi:10.1134/S2079978011030010.
- ↑ Wayiza Masamba: Petasis vs. Strecker Amino Acid Synthesis: Convergence, Divergence and Opportunities in Organic Synthesis. In: Molecules. Band 26, Nr. 6, 18. März 2021, S. 1707, doi:10.3390/molecules26061707, PMID 33803879, PMC 8003338 (freier Volltext).
- ↑ Jarugu Narasimha Moorthy, Nidhi Singhal: Facile and Highly Selective Conversion of Nitriles to Amides via Indirect Acid-Catalyzed Hydration Using TFA or AcOH−H 2 SO 4. In: The Journal of Organic Chemistry. Band 70, Nr. 5, 1. März 2005, S. 1926–1929, doi:10.1021/jo048240a.
- ↑ Manas K Basu, Fen-Tair Luo: Efficient transformation of nitrile into amide under mild condition. In: Tetrahedron Letters. Band 39, Nr. 19, Mai 1998, S. 3005–3006, doi:10.1016/S0040-4039(98)00444-4.
- ↑ Charles R. Hauser, David S. Hoffenberg: CONVERSION OF NITRILES TO AMIDES AND ACIDS BY MEANS OF BORON FLUORIDE. In: The Journal of Organic Chemistry. Band 20, Nr. 10, Oktober 1955, S. 1448–1453, doi:10.1021/jo01127a025.
- ↑ Kazuya Yamaguchi, Mitsunori Matsushita, Noritaka Mizuno: Efficient Hydration of Nitriles to Amides in Water, Catalyzed by Ruthenium Hydroxide Supported on Alumina. In: Angewandte Chemie International Edition. Band 43, Nr. 12, 12. März 2004, S. 1576–1580, doi:10.1002/anie.200353461.
- ↑ N. Naresh Kumar Reddy, Sadu Nageswara Rao, Rajendra D. Patil, Subbarayappa Adimurthy: Transition metal-free hydration of nitriles to amides mediated by NaOH. In: Advanced Material Science. Band 3, Nr. 1, 2018, doi:10.15761/AMS.1000137.
- ↑ Shreenath Prasad, Tek Chand Bhalla: Nitrile hydratases (NHases): at the interface of academia and industry. In: Biotechnol Adv. Band 28, Nr. 6, 2010, S. 725-41, doi:10.1016/j.biotechadv.2010.05.020.
- ↑ Bo Wu, Ji Zhang, Meng Yang, Yang Yue, Li-Jian Ma, Xiao-Qi Yu: Raney Ni/KBH4: An efficient and mild system for the reduction of nitriles to amines. In: Arkivoc. Band 2008, Nr. 12, 4. April 2008, S. 95–102, doi:10.3998/ark.5550190.0009.c11.
- ↑ Shankare Gowda, D.Channe Gowda: Application of hydrazinium monoformate as new hydrogen donor with Raney nickel: a facile reduction of nitro and nitrile moieties. In: Tetrahedron. Band 58, Nr. 11, März 2002, S. 2211–2213, doi:10.1016/S0040-4020(02)00093-5.
- ↑ P Schärdinger, T Muller, J Lercher: Investigations into the mechanism of the liquid-phase hydrogenation of nitriles over Raney-Co catalysts. In: Journal of Catalysis. Band 253, Nr. 1, 1. Januar 2008, S. 167–179, doi:10.1016/j.jcat.2007.10.008.
- ↑ Atsuko Nose, Tadahiro Kudo: Studies of reduction with dimethoxyborane-transition metal boride systems. In: Chemical and Pharmaceutical Bulletin. Band 38, Nr. 6, 1990, S. 1720–1723, doi:10.1248/cpb.38.1720.
- ↑ Morris Freifelder: A Low Pressure Process for the Reduction of Nitriles. Use of Rhodium Catalyst 1. In: Journal of the American Chemical Society. Band 82, Nr. 9, Mai 1960, S. 2386–2389, doi:10.1021/ja01494a067.
- ↑ R Ramachandran: An overview of industrial uses of hydrogen. In: International Journal of Hydrogen Energy. Band 23, Nr. 7, Juli 1998, S. 593–598, doi:10.1016/S0360-3199(97)00112-2.
- ↑ Siegfried R. Waldvogel: Strategic Applications of Named Reactions in Organic Synthesis. Background and Detailed Mechanisms. By Laszlo Kürti and Barbara Czako. In: Angewandte Chemie International Edition. Band 44, Nr. 32, 8. August 2005, S. 5005–5006, doi:10.1002/anie.200585304.
- ↑ Franz J. Weiberth, Stan S. Hall: Copper(I)-activated addition of Grignard reagents to nitriles. Synthesis of ketimines, ketones, and amines. In: The Journal of Organic Chemistry. Band 52, Nr. 17, August 1987, S. 3901–3904, doi:10.1021/jo00226a033.
- ↑ H. Surya Prakash Rao, Shaik Rafi, K. Padmavathy: The Blaise reaction. In: Tetrahedron. Band 64, Nr. 35, August 2008, S. 8037–8043, doi:10.1016/j.tet.2008.05.109.
- ↑ Adolf Pinner: Die Imidoäther und ihre Derivate. Robert Oppenheim, 1892, ISBN 978-0-598-98239-1.
- ↑ Andriani G. Chaidali, Ioannis N. Lykakis: Synthetic Routes to Imidates and Their Applications in Organic Transformation: Recent Progress. In: European Journal of Organic Chemistry. Band 26, Nr. 39, 16. Oktober 2023, doi:10.1002/ejoc.202300497.
- ↑ Kiyoshi Watanabe, Naoto Kogoshi, Hideaki Miki, Yasuhiro Torisawa: Improved Pinner Reaction with CPME as a Solvent. In: Synthetic Communications. Band 39, Nr. 11, 7. Mai 2009, S. 2008–2013, doi:10.1080/00397910802632548.
- ↑ Richard R. Schmidt, Josef Michel: Facile Synthesis of α‐ and β‐ O ‐Glycosyl Imidates; Preparation of Glycosides and Disaccharides. In: Angewandte Chemie International Edition in English. Band 19, Nr. 9, September 1980, S. 731–732, doi:10.1002/anie.198007311.
- ↑ You Yang, Xiaheng Zhang, Biao Yu: O-Glycosylation methods in the total synthesis of complex natural glycosides. In: Natural Product Reports. Band 32, Nr. 9, 2015, S. 1331–1355, doi:10.1039/C5NP00033E.
- ↑ Rodney A. Fernandes, Pullaiah Kattanguru, Sachin P. Gholap, Dipali A. Chaudhari: Recent advances in the Overman rearrangement: synthesis of natural products and valuable compounds. In: Organic & Biomolecular Chemistry. Band 15, Nr. 13, 2017, S. 2672–2710, doi:10.1039/C6OB02625G.
- ↑ Daisuke Uraguchi, Ryosuke Tsutsumi, Takashi Ooi: Catalytic asymmetric Payne oxidation under the catalysis of P-spiro chiral triaminoiminophosphorane: application to the synthesis of N-sulfonyl oxaziridines. In: Tetrahedron. Band 70, Nr. 8, Februar 2014, S. 1691–1701, doi:10.1016/j.tet.2013.12.086.
- ↑ George B. Payne, Philip H. Deming, Paul H. Williams: Reactions of Hydrogen Peroxide. VII. Alkali-Catalyzed Epoxidation and Oxidation Using a Nitrile as Co-reactant. In: The Journal of Organic Chemistry. Band 26, Nr. 3, März 1961, S. 659–663, doi:10.1021/jo01062a004.
- ↑ Fan Xu, Jianhua Sun, Qi Shen: Samarium diiodide promoted synthesis of N,N ′-disubstituted amidines. In: Tetrahedron Letters. Band 43, Nr. 10, März 2002, S. 1867–1869, doi:10.1016/S0040-4039(02)00129-6.
- ↑ John H. Forsberg, Vincent T. Spaziano, Trichey M. Balasubramanian, Gordon K. Liu, Steven A. Kinsley, Charles A. Duckworth, John J. Poteruca, Paul S. Brown, Judith L. Miller: Use of lanthanide(III) ions as catalysts for the reactions of amines with nitriles. In: The Journal of Organic Chemistry. Band 52, Nr. 6, März 1987, S. 1017–1021, doi:10.1021/jo00382a009.
- ↑ Frederick G. Bordwell, Herbert E. Fried: Acidities of the hydrogen-carbon protons in carboxylic esters, amides, and nitriles. In: The Journal of Organic Chemistry. Band 46, Nr. 22, Oktober 1981, S. 4327–4331, doi:10.1021/jo00335a001.
- ↑ Victor A. Lucas-Rosales, Marco A. García-Revilla, J. Oscar C. Jiménez-Halla: Computational Revision of the Mechanism of the Thorpe Reaction. MDPI, 14. November 2022, S. 29, doi:10.3390/ecsoc-26-13550.
- ↑ Yasuo Sato, Michihisa Yato, Tomohiko Ohwada, Shinichi Saito, Koichi Shudo: Involvement of Dicationic Species as the Reactive Intermediates in Gattermann, Houben-Hoesch, and Friedel-Crafts Reactions of Nonactivated Benzenes. In: Journal of the American Chemical Society. Band 117, Nr. 11, März 1995, S.